Pathophysiology of Pulmonary Hypertension
Marlene Rabinovitch
Rachel K. Hopper
Introduction
This chapter addresses the pathophysiologic mechanisms leading to pulmonary hypertension associated with congenital heart defects and other disease-related or idiopathic processes. The new classification of pulmonary hypertension adopted by the Fifth World Symposium on Pulmonary Hypertension is included in Table 65.1 (1). Our evolving knowledge of fundamental mechanisms has led to new and improved treatments and holds great promise for the development of future therapies.
TABLE 65.1 Updated Classification of Pulmonary Hypertension | ||||||
|---|---|---|---|---|---|---|
|
Heart Defects with Increased Pulmonary Blood Flow
High-Flow Congenital Heart Defects and Pulmonary Hypertension
As early as 1935, Brenner (2) observed different types of pulmonary vascular lesions in patients with congenital heart defects. A variety of physiologic factors appeared to contribute to the severity and rate at which vascular disease developed in patients with congenital heart defects (Table 65.2). In 1958, Heath and Edwards (3) suggested that there was a progression of structural changes (grades I through VI) (Fig. 65.1). Grades I and II, medial hypertrophy and intimal hyperplasia, respectively, were considered mild and probably reversible. Grades III and IV were, respectively, lumen occlusion from intimal hyperplasia and early to advanced arterial dilation. Grade III was thought to be, at best, partially reversible, but grade IV, irreversible. Grades V and VI were considered terminal changes, V being angiomatoid formation and VI fibrinoid necrosis. The term plexogenic arteriopathy or plexiform lesions has been adopted to refer to those occlusive and dilatation lesions containing expansion of smooth muscle- and endothelial-like cells. The changes beyond grades I–III were identified most frequently in older children. Structural changes that predicted severe and fixed elevation in pulmonary vascular resistance were therefore more difficult to establish in infants and young children, although it was recognized that certain lesions, such as transposition of the great arteries with intact septum, but especially with a large ventricular septal defect (VSD) or a patent ductus arteriosus, were at high risk. Curiously, there have been reports of late progression of pulmonary vascular disease in infants with transposition of the great arteries even after successful and timely surgical correction. Infants with a large VSD, especially an atrioventricular septal defect, also developed pulmonary vascular disease in early infancy; in the latter group, there were advanced changes observed even within the first 6 months of life. Patients with cyanotic heart defects who had large, surgically created systemic-to-pulmonary artery shunts were also at risk for pulmonary vascular disease. Several investigators tried to quantify the degree of medial hypertrophy, but their measurements did not correlate closely with the preoperative level of pulmonary vascular resistance or with its change postoperatively (4).
Structural Remodeling and Growth
Beginning in 1965, a new and quantitative method of analysis of the pulmonary vascular bed was developed that was particularly applicable to the study of infants and young children because it incorporated features of altered growth of the pulmonary circulation. This method was based upon extensive studies carried out using a technique of radiopaque barium–gelatin arterial injection of a postmortem lung specimen. It was observed from the postmortem arteriograms that the vessels are prominent in the newborn, whereas in the adult they are obscured by a dense background haze produced by the addition of many small intra-acinar arteries not present at birth (5). On microscopic examination, three features of normal remodeling and growth of the pulmonary vascular bed were established (6). With increasing age, muscle is observed in arteries located more peripherally within the acinus. At first, nonmuscular arteries become partially muscular, and later they become fully muscularized. At birth, the muscularized arteries are thick walled, but within a few days, the smallest muscular arteries dilate, and their walls thin to adult levels. When infants are 4 months of age, this process has included the largest muscular pulmonary arteries and is complete. Arteries grow both in number and size, and they grow most rapidly in infancy. Although alveoli also proliferate, the ratio of alveoli to arteries actually decreases from the newborn value of 20:1 to the value of 8:1, which is achieved first in early childhood and which persists (Fig. 65.2).
TABLE 65.2 Factors Determining the Development of Pulmonary Vascular Disease in Patients with Common Varieties of Congenital Heart Disease | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
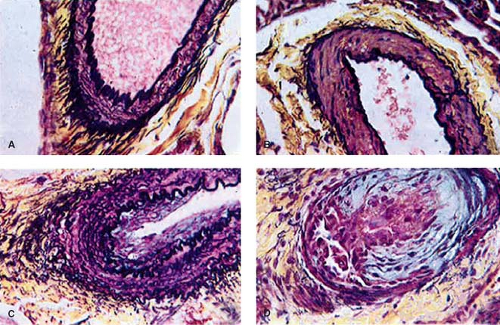 Figure 65.1 Progressive changes as described by Heath and Edwards: Movat pentachrome staining of lung sections from patients with pulmonary arterial changes. In (A), is a pulmonary artery with mild medial hypertrophy. In (B) the medial hypertrophy is more severe (Heath–Edwards grade 1). C: Shows a vessel with eccentric neointimal encroaching on the lumen (Heathe–Edwards grade II). D: Shows a plexiform lesion with an occlusive neointima, a thinned out media, loss of elastin and evidence of neoangiogenic channels (grade IV). Grade III (not shown) would be similar to (D) but with an intact media and no evidence of neoangiogenesis in the lumen. (From Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150(4):1349–1360.) |
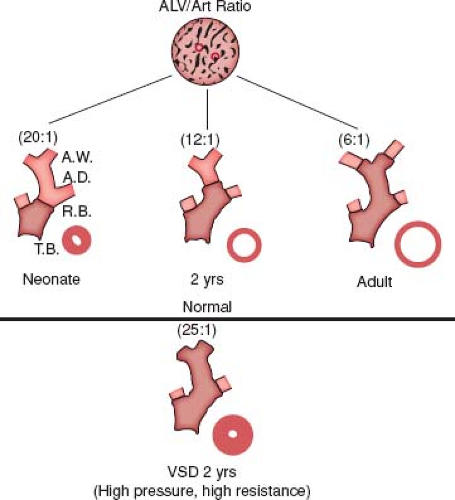 Figure 65.2 Schema showing peripheral pulmonary arterial development through morphometric changes: extension of muscle into peripheral arteries, percent wall thickness, and artery number (alveolar–arterial ratio) as they relate to age. Upper panel: Normal development. Bottom panel: Abnormalities in all three features in a 2-year-old child with a hypertensive VSD. T.B., artery accompanying a terminal bronchiolus; R.B., artery accompanying a respiratory bronchiolus; A.D., artery accompanying an alveolar duct; A.W., artery accompanying an alveolar wall; ALV/Art, alveolar–arterial ratio. (From Rabinovitch M, Haworth SG, Castaneda AR, Nadas AS, Reid LM. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation. 1978;58:1107–1122, with permission.) |
TABLE 65.3 Structural Features Quantified in Congenital Heart Defects | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Morphometric analysis of the lungs of patients with congenital heart defects revealed disturbed growth and remodeling of the pulmonary vascular bed (Table 65.3) (7). On postmortem arteriograms from infants with VSD, the axial arteries were dilated at the hilum but narrowed peripherally. On microscopic examination of the pulmonary vascular bed, muscle had extended precociously into normally nonmuscular peripheral arteries, regression of the perinatal musculature had not occurred, and there was additional medial hypertrophy. Also, the peripheral arteries had not grown normally in that they were small and few. Alveolar differentiation and multiplication, however, were normal. Because there were no regional variations in the lung, it was feasible to apply the morphometric technique to analysis of lung biopsy tissue in assessing abnormal muscularity and evaluating arterial size and number (8).
Lung Biopsy
Lung biopsy studies indicated that the severity of altered growth and development of the pulmonary vascular bed correlates with the hemodynamic state. Three progressively severe stages are seen. With grade A, there is abnormal extension of muscle into small peripheral arteries, or, in addition, there is a mild increase in wall thickness of the normally muscular arteries (<1.5 times normal). These patients have increased pulmonary blood flow and increased pulse pressure but normal mean pulmonary artery pressure. Meyrick and Reid (9) showed from ultrastructural studies of lung biopsy tissue that this change is due to precocious differentiation of pericytes to mature smooth muscle cells in the nonmuscular region of the artery and the intermediate cell in the partially muscular region. Because arteries become more muscular as they increase in size, it is tempting to speculate that, in the altered hemodynamic setting of chronic high flow and high pressure, “stretch” is the stimulus for smooth muscle cell differentiation from precursor cells.
With grade B, as in grade A, there is increased extension of muscle, but, in addition, there is more severe medial hypertrophy of normally muscular arteries. When medial wall thickness is greater than 1.5 but less than 2 times normal (mild grade B), pulmonary hypertension is usually present; when medial wall thickness is more than twice normal (severe grade B), pulmonary hypertension is invariably present and usually the pressure value is greater than half that of the systemic level. The medial thickness is due to hypertrophy as well as hyperplasia of pre-existing smooth muscle cells and also to an increase in the intercellular connective tissue proteins.
With grade C, in addition to the findings of severe grade B, arterial concentration and usually artery size are reduced. Patients with these changes generally have a pulmonary vascular resistance of greater than 3.5 U·m2. When the artery number is less than half of normal (severe grade C), pulmonary vascular resistance values are often in excess of 6 U·m2. The basis for grade C is likely the failure of new vessels to grow normally, although some loss of arteries also may occur. Morphometric grades A and B are refinements of Heath–Edwards grade I; grade C is a new feature that also appears to be of important functional significance. Grade C may be found with Heath–Edwards grade I, is common with grade II, and is invariable with grade III. In fact, when grade III is seen, arterial concentration is generally half of normal or less.
Whether and to what extent abnormal growth and structural remodeling of the pulmonary vascular bed are permanent and result in persistent pulmonary hypertension was estimated by correlating these features with postoperative hemodynamic studies. The quantitative features of abnormal growth and remodeling of the pulmonary arteries and the qualitative changes described by Heath and Edwards correlate with the hemodynamic behavior of the pulmonary circulation in the immediate postoperative period in the intensive care unit 1 day after repair and at the time of routine cardiac catheterization study 1 year later (10). Patients with grade A or mild grade B changes have normal pulmonary artery pressures in the early postoperative period or only a minimal degree of elevation. Most patients with more severe medial hypertrophy (i.e., severe grade B and Heath–Edwards I) have elevated values. The pulmonary hypertension is frequently labile and almost always can be controlled; the mechanism and treatment are discussed in the next section. Both the presence and the severity of pulmonary hypertension in the early postoperative period are increasingly predictable when there are more advanced changes on lung biopsy, that is, reduced artery number (grade C) and intimal hyperplasia (Heath–Edwards II and III) (Fig. 65.3).
One year after repair, however, patients operated within the first 8 months of life tend to have normal pulmonary hemodynamics regardless of the severity of vascular changes on lung biopsy, as do patients with severe grade B (Heath–Edwards I) abnormalities, regardless of their age at repair. Patients surgically corrected between 9 months and 2 years of life with grade C and Heath–Edwards II or III structural changes may have persistent elevation in pulmonary vascular resistance; this appears inevitable in patients operated after the age of 2 years (Fig. 65.3).
While not often used clinically in the current era, quantitative techniques have been applied successfully to the analysis of lung biopsy tissue prepared by frozen section to aid decision making between a palliative or corrective procedure when preoperative hemodynamic data are borderline or difficult to obtain or to interpret. The ability to predict whether even mild elevation in pulmonary vascular resistance will be present postoperatively is of
increasing importance, particularly in the consideration of patients for a Fontan procedure (11). Patients with tricuspid atresia who have had previous systemic-to-pulmonary artery shunts and those with single-ventricle and previous pulmonary artery bands are at particular risk. We consider a biopsy showing severe grade C (less than half the normal number of arteries) and grade III or greater changes in 20% of vessels to indicate severe vascular disease that is unlikely to regress postoperatively and may preclude a successful result from closure of a VSD; severe grade B, medial wall thickness greater than twice normal, or grade II changes in any vessel may preclude a favorable result from a Fontan procedure. Even minor vascular changes on lung biopsy tissue (mild grade B) are associated with increased morbidity after the Fontan procedure, as gauged by prolonged hospitalization; the need for increased ventilator support; and drainage from chest tubes (unpublished observations). This highlights the need for more sensitive noninvasive techniques to determine the presence of pulmonary vascular disease in this population.
increasing importance, particularly in the consideration of patients for a Fontan procedure (11). Patients with tricuspid atresia who have had previous systemic-to-pulmonary artery shunts and those with single-ventricle and previous pulmonary artery bands are at particular risk. We consider a biopsy showing severe grade C (less than half the normal number of arteries) and grade III or greater changes in 20% of vessels to indicate severe vascular disease that is unlikely to regress postoperatively and may preclude a successful result from closure of a VSD; severe grade B, medial wall thickness greater than twice normal, or grade II changes in any vessel may preclude a favorable result from a Fontan procedure. Even minor vascular changes on lung biopsy tissue (mild grade B) are associated with increased morbidity after the Fontan procedure, as gauged by prolonged hospitalization; the need for increased ventilator support; and drainage from chest tubes (unpublished observations). This highlights the need for more sensitive noninvasive techniques to determine the presence of pulmonary vascular disease in this population.
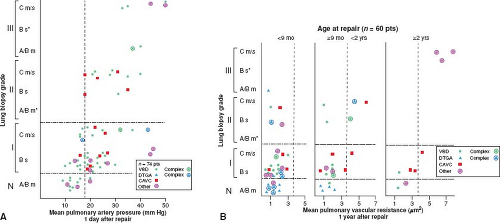 Figure 65.3 A: Lung biopsy grade is correlated with mean pulmonary arterial pressure recorded the day after surgical repair. The dashed vertical lines separate the normal from the abnormally elevated pressure values, and the dotted horizontal lines separate the biopsy grades. Note that with the more severe Heath–Edwards changes on lung biopsy tissue, there is a trend toward a greater proportion of patients with elevated pulmonary arterial pressures and higher values. A, B, C, morphometric grades; m, mild, s, severe; N, I, II, III, Heath–Edwards grades; n, normal; *, no patients in this group; VSD, ventricular septal defect; DTGA, d-transposition of the great arteries; CAVC, complete artioventricular canal; complex, associated abnormality. B: Graph correlating lung biopsy grade with pulmonary vascular resistance 1 year after cardiac repair. Patients who underwent repair within the first 8 months of life, but not those operated on later, had normal pulmonary vascular resistance regardless of the severity of their structural changes. (From Rabinovitch M, Keane JP, Norwood WI, Castaneda AR, Reid L. Vascular structure in lung biopsy tissue correlated with pulmonary hemodynamic findings after repair of congenital heart defects. Circulation. 1984;69:655–667.) |
Wedge Angiography
Techniques of wedge angiography have been developed to assess preoperatively the structural state of the pulmonary vascular bed. Changes that can be evaluated quantitatively, such as sparsity of arborization of the pulmonary tree, abrupt termination, tortuosity and narrowing of small arteries, and reduced background capillary filling, generally reflect advanced changes in the preacinar arteries of at least Heath–Edwards grade III severity. A technique has been described that allows quantitative assessment of abnormalities in a pulmonary wedge angiogram (12). A balloon catheter is directed to the origin of the axial artery of the posterior basal segment of the right lower lobe; contrast material is injected, and the injection is filmed on biplane cineangiography. The rate of tapering of the arteries is assessed by measuring the length of a segment over which the lumen diameter narrows from 2.5 mm to 1.5 mm. More abrupt arterial tapering is suggestive of more severe changes in the intra-acinar arteries, assessed both morphometrically and by the Heath–Edwards classification (Table 65.4 and Fig. 65.4).
TABLE 65.4 Correlation of the Pathology, Physiology, and Angiogram in Pulmonary Vascular Disease | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
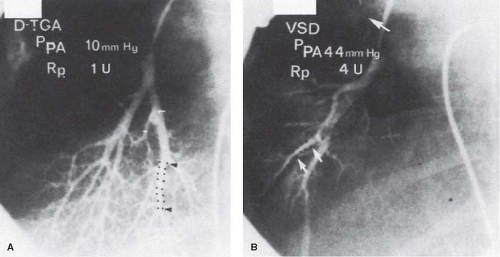 Figure 65.4 A: A wedge angiogram shows slow tapering of the axial artery in a child with TGA and normal pulmonary artery pressure (PPA) and resistance (Rp). Approximate segment length between 2.5 and 1.5 mm internal diameter is marked off (arrowheads). B: A wedge angiogram in a child with a VSD shows rapid tapering of the artery when there is increased PPA and Rp. An approximate segment length between 2.5 and 1.5 mm internal diameter is marked off (arrows). Large arrow denotes takeoff to the right pulmonary artery. (From Rabinovitch M, Reid L. Quantitative structural analysis of the pulmonary vascular bed in congenital heart defects. In: Engle MA, ed. Cardiovascular Clinics: Pediatric Cardiovascular Disease. Philadelphia, PA: FA Davis; 1981, with permission.) |
Further studies revealed that there are several pitfalls and limitations in the interpretation of the pulmonary wedge angiogram. Pulmonary stenosis or previous placement of a pulmonary artery band, owing to poststenotic dilation, will give the impression of rapid tapering. With advanced vascular disease, there is sometimes such extensive intimal hyperplasia that the vessel appears narrowed all the way from the hilum so that abrupt tapering is no longer apparent. In this situation, however, the background haze is absent and the pulmonary circulation time is usually prolonged. Technical abnormalities also can result in difficulties in interpretation. If the injection of contrast fails to fill the vessels all the way out to the pleura, the background will appear dark, and if the balloon does not completely occlude the vessel, the false impression of a dense background will be created owing to filling of capillaries and veins. Assessment of the circulation time depends on the exclusion of pulmonary vein stenosis and intrapulmonary shunting.
Intravascular ultrasound could be investigated to estimate the severity of pulmonary vascular disease at least in proximal vessels. Optical coherence tomography is a new technique that can resolve neointimal lesions in very small arteries and may hold greater promise, but has just recently been applied clinically to assess coronary stent stenosis (13). Early studies of optical coherence tomography in the pulmonary vasculature studies have shown correlation in pulmonary artery remodeling with changes in histology (14).
Reactive Pulmonary Circulation
A major challenge in pediatric cardiology is to understand and control the reactive pulmonary circulation, which can be particularly problematic in the early postoperative period. The pulmonary hypertensive crisis, as it has been called, is thought to result from interaction of a hypertrophied and perhaps hypercontractile circulation with an injured vascular endothelium, with platelets and leukocytes that were exposed to postcardiopulmonary bypass and hypothermia and may more easily degranulate and release potent vasoconstrictor agents, particularly thromboxanes and leukotrienes. In recent studies, increased density of neuroepithelial bodies has been observed in the airways of patients at risk for this complication (15). The neuroendocrine cells, which are also oxygen sensors, contain bombesin and serotonin, agents known to be potent vasoconstrictors. There is also an increase in vasoconstrictor neuropeptide-containing nerves (16) (Fig. 65.5). Because most of the pulmonary hypertensive crises occur while weaning from the ventilator, it is tempting to speculate that swings in airway pressure might lead to degranulation of the neuroepithelial cells and release of the vasoconstrictor substances. Moreover, there is a striking decrease in lung compliance accompanying the pulmonary hypertensive crisis.
In ultrastructural lung biopsy studies from patients with congenital heart defects and pulmonary hypertension, alterations in endothelial cells support endothelial dysfunction as a cause of heightened pulmonary vascular reactivity and also relate endothelial dysfunction to the pathogenesis of progressive pulmonary vascular disease. On scanning electron microscopy, the endothelial surface of normal thin-walled pulmonary arteries has a “corduroy-like” appearance in that the cells form narrow, even ridges. In contrast, the endothelial surface of hypertensive thick-walled pulmonary arteries has a “cable-like” texture in that the cells form deep, twisted ridges. The hypertensive endothelium may be predisposed to interact abnormally with marginating blood elements, such as platelets and leukocytes. This might result in the release of pulmonary vasoconstrictor substances and smooth muscle mitogens (17).
On transmission electron microscopy, the endothelium appeared to show heightened metabolic activity with an increased rough endoplasmic reticulum. The subendothelium of the muscular arteries is also abnormal in that there appears to be degradation and neosynthesis of the internal elastic lamina. This observation provided an important clue related to the discovery of heightened elastolytic activity in the vessel wall associated with the initiation and progression of pulmonary vascular disease. Further studies were designed
to try to identify products of increased endothelial metabolism. Since endothelial cells produce von Willebrand factor (vWF, factor VIII), an increase in this protein was proposed to cause platelet adhesion and formation of microthrombi and the release of pulmonary vasoconstrictor substances and smooth muscle mitogens. Using an immunoperoxidase stain for factor VIII, hypertensive pulmonary arteries stained densely, whereas nonhypertensive vessels did not. Circulating levels of the factor VIII molecule, both the antigenic component (VIII:Ag) and the biologic activity, assessed as ristocetin-induced platelet agglutination (VIII:rist) were measured. Although VIII:Ag levels were significantly higher in patients with congenital heart defects and elevated pulmonary artery pressure than in those with normal pressure, only a few patients showed a concomitant elevation in biologic activity. This finding indicated that the molecule being secreted was lacking in biologic activity; indeed, further biochemical analysis of the factor VIII molecule showed lack of the high–molecular-weight components (18). However, under conditions that aggravate endothelial dysfunction (e.g., cardiopulmonary bypass), vWF biologic activity was markedly increased (19). This could indeed account for the development of platelet fibrin microthrombi in the postoperative period and for abnormal release of vasoactive compounds causing increased vascular reactivity. Various regimens have been adopted to anticipate and manage postoperative increased pulmonary vascular reactivity, which include monitoring pulmonary artery and left atrial pressures, institution of nitric oxide (NO) therapy, and the institution of phosphodiesterase inhibitors such as sildenafil, if therapy is needed beyond the intensive care unit.
to try to identify products of increased endothelial metabolism. Since endothelial cells produce von Willebrand factor (vWF, factor VIII), an increase in this protein was proposed to cause platelet adhesion and formation of microthrombi and the release of pulmonary vasoconstrictor substances and smooth muscle mitogens. Using an immunoperoxidase stain for factor VIII, hypertensive pulmonary arteries stained densely, whereas nonhypertensive vessels did not. Circulating levels of the factor VIII molecule, both the antigenic component (VIII:Ag) and the biologic activity, assessed as ristocetin-induced platelet agglutination (VIII:rist) were measured. Although VIII:Ag levels were significantly higher in patients with congenital heart defects and elevated pulmonary artery pressure than in those with normal pressure, only a few patients showed a concomitant elevation in biologic activity. This finding indicated that the molecule being secreted was lacking in biologic activity; indeed, further biochemical analysis of the factor VIII molecule showed lack of the high–molecular-weight components (18). However, under conditions that aggravate endothelial dysfunction (e.g., cardiopulmonary bypass), vWF biologic activity was markedly increased (19). This could indeed account for the development of platelet fibrin microthrombi in the postoperative period and for abnormal release of vasoactive compounds causing increased vascular reactivity. Various regimens have been adopted to anticipate and manage postoperative increased pulmonary vascular reactivity, which include monitoring pulmonary artery and left atrial pressures, institution of nitric oxide (NO) therapy, and the institution of phosphodiesterase inhibitors such as sildenafil, if therapy is needed beyond the intensive care unit.
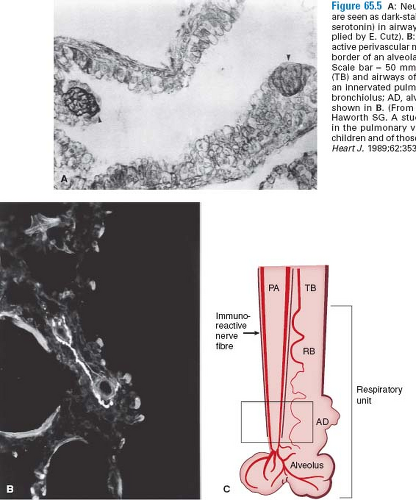 Figure 65.5 A: Neuroepithelial bodies (arrowheads) are seen as dark-staining regions (immunoreactive for serotonin) in airway of a newborn infant (Kindly supplied by E. Cutz). B: Tyrosine hydroxylase immunoreactive perivascular nerve fibers at the advential–medial border of an alveolar duct artery in a child aged 2 1/2. Scale bar = 50 mm. C: Shows terminal bronchiolus (TB) and airways of respiratory unit accompanied by an innervated pulmonary artery (PA). RB, respiratory bronchiolus; AD, alveolar duct. Square indicates area shown in B. (From Allen KM, Wharton J, Polak JM, Haworth SG. A study of nerves containing peptides in the pulmonary vasculature of healthy infants and children and of those with pulmonary hypertension. Br Heart J. 1989;62:353–360, with permission.) |
There is now convincing evidence that impaired production of NO appears at an early time point in patients with congenital heart defects (20). There also is evidence that production of the vasoconstrictor endothelin also might be increased in patients with pulmonary hypertension and congenital heart defects (21).
Many patients with high-flow congenital heart defects that are operated upon in a timely fashion show a fall in pulmonary artery pressure and return to normal resting hemodynamics, indicating resolution and regression of pulmonary hypertensive structural changes. This is supported by experimental studies and by anecdotal reports of resolution of severe pulmonary vascular disease in the remaining lung after single lung transplant. There are, however, some patients who maintain a high level of pulmonary vascular resistance and are refractory to vasodilator therapy despite what appear to be mild vascular changes on light microscopy (medial hypertrophy),
and others who develop rapidly progressive pulmonary vascular disease despite early diagnosis and timely intervention. For these patients, the prognosis may be not much better than those with unexplained pulmonary hypertension (22). A recent study of outcomes of patients with congenital heart disease (systemic-to-pulmonary shunt lesions) and pulmonary hypertension in the current treatment era demonstrated significantly decreased 20-year survival in patients with residual pulmonary vascular disease following defect closure (36%) relative to patients with Eisenmenger syndrome (87%) or those patients with an unrepaired systemic-to-pulmonary shunt without Eisenmenger syndrome (86%). While preoperative hemodynamics are unknown for the surgically repaired group, the late average age at operation may suggest advanced pulmonary vascular disease at the time of operation and inability of the right ventricle to adapt to the increased workload (23). Considerations regarding operability are discussed in Chapter 66.
and others who develop rapidly progressive pulmonary vascular disease despite early diagnosis and timely intervention. For these patients, the prognosis may be not much better than those with unexplained pulmonary hypertension (22). A recent study of outcomes of patients with congenital heart disease (systemic-to-pulmonary shunt lesions) and pulmonary hypertension in the current treatment era demonstrated significantly decreased 20-year survival in patients with residual pulmonary vascular disease following defect closure (36%) relative to patients with Eisenmenger syndrome (87%) or those patients with an unrepaired systemic-to-pulmonary shunt without Eisenmenger syndrome (86%). While preoperative hemodynamics are unknown for the surgically repaired group, the late average age at operation may suggest advanced pulmonary vascular disease at the time of operation and inability of the right ventricle to adapt to the increased workload (23). Considerations regarding operability are discussed in Chapter 66.
Therapies for the patient with Eisenmenger syndrome have included chronic oxygen, anticoagulants, and palliative surgical procedures, including atrial septal defect creation and intravenous as well as, more recently, subcutaneous, inhaled, or oral prostacyclin analogs. In some cases, these measures have improved the quality of life and in others they have acted as a bridge to a heart–lung transplant or surgical correction along with lung transplant. More recent addition of phosphodiesterase inhibitors and endothelin receptor antagonists in this group of patients awaits the results of clinical trials, but is discussed later in this chapter, and in the subsequent chapter, with regard to patients with idiopathic pulmonary hypertension.
Pathophysiology Based Upon Further Pathologic Assessments
Recent immunohistochemical studies have been carried out in lung biopsy tissue from patients with congenital heart defects to elucidate mechanisms that are directly related to enhanced proliferation and migration of cells in the neointima with characteristics of smooth muscle cells. There is a progressive increase in the deposition of two matrix glycoproteins, tenascin and fibronectin, in the media and neointima (Fig. 65.6). We previously related the increased expression of tenascin to vascular smooth muscle cells
and fibroblasts. Fibronectin has been related to increased migration of smooth muscle-like cells in the context of neointimal formation. There is evidence from other studies that the neointimal lesions are associated with increased expression of transforming growth factor-β (TGF-β) and procollagen in addition to fibronectin (24). It is also proposed that endothelial cell proliferation and a form of angiogenesis is observed with plexiform lesions. The plexiform lesions in pulmonary hypertension associated with congenital heart disease appear to be derived from different clonal populations of endothelial cells compared with those observed in unexplained pulmonary hypertension where a single clone is usually found (25). More recently, an increase in the expression of the calcium binding protein S100A4/Mts1 was reported in advanced lesions from patients with congenital heart defects causing pulmonary arterial hypertension (PAH) as well as idiopathic PAH (26) (Fig. 65.7). S100A4/Mts1 stimulates vascular smooth muscle cell migration and proliferation, is produced in response to serotonin stimulation (27) and is enhanced when there is increased activity of the serotonin transporter (SERT). This is in keeping with studies indicating that a polymorphism causing increased activity of the SERT is highly prevalent in patients with PAH (28).
and fibroblasts. Fibronectin has been related to increased migration of smooth muscle-like cells in the context of neointimal formation. There is evidence from other studies that the neointimal lesions are associated with increased expression of transforming growth factor-β (TGF-β) and procollagen in addition to fibronectin (24). It is also proposed that endothelial cell proliferation and a form of angiogenesis is observed with plexiform lesions. The plexiform lesions in pulmonary hypertension associated with congenital heart disease appear to be derived from different clonal populations of endothelial cells compared with those observed in unexplained pulmonary hypertension where a single clone is usually found (25). More recently, an increase in the expression of the calcium binding protein S100A4/Mts1 was reported in advanced lesions from patients with congenital heart defects causing pulmonary arterial hypertension (PAH) as well as idiopathic PAH (26) (Fig. 65.7). S100A4/Mts1 stimulates vascular smooth muscle cell migration and proliferation, is produced in response to serotonin stimulation (27) and is enhanced when there is increased activity of the serotonin transporter (SERT). This is in keeping with studies indicating that a polymorphism causing increased activity of the SERT is highly prevalent in patients with PAH (28).
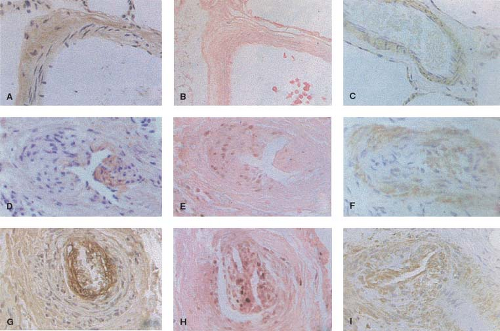 Figure 65.6 Representative photomicrographs showing immunoperoxidase staining for tenascin-C (TN) (A, D, G), proliferating cell nuclear antigen (PCNA) (B, E, H), and epidermal growth factor (EGF) (C, F, I) in graded lung biopsy tissue sections. A, B, C: Vessel showing a typical grade IA lesion; D, E, F: Vessel showing a typical grade IC lesion; G, H, I: Vessel showing a typical grade IIIC lesion. In low-grade lesions (A), modest TN immunostaining was evident in the adventitia. With medial hypertrophy, TN immunoreactivity became more prominent in the periendothelium (D), with the most intense immunostaining being apparent within the neointima of high-grade lesions showing occlusive neointimal formation (G). In the lowest grade of lesion, PCNA was negative (B), despite foci of EGF in the media. With medial hypertrophy, PCNA was expressed in the media (E), together with foci of EGF (F). With the development of higher-grade occlusive lesions, TN (G), PCNA (H), and EGF (I) colocalized to the neointimal cell layers. Note that TN and PCNA staining was performed on serial sections, whereas EGF detection was carried out on similar vessels within the same biopsy. Original magnification, ×40. (From Jones PL, Cowan KN, Rabinovitch. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150:1349–1360, with permission.) |
Experimental Studies Simulating High Flow and Pressure
In experimental studies, an aortopulmonary shunt surgically created in a growing piglet results in a progressive increase in pulmonary artery pressure associated with the development of structural changes similar to those seen in the clinical setting. Creation of large aortopulmonary shunts in dogs, particularly into a single pulmonary artery, resulted in more rapidly progressive pulmonary vascular changes. There are several other experimental models of high-flow congenital heart defects such as sheep or calves after an aortopulmonary lobar anastomosis. In utero placement of an aortopulmonary shunt most faithfully reproduces the changes that might be expected to occur in the newborn with a left-to-right shunt when the pulmonary vascular resistance falls. In this model, the initial response to high flow induced from the time of birth experimentally appears to be angiogenic with high levels of vascular endothelial growth factor (VEGF) and its receptors, but later, in association with the decrease in VEGF and decline in arterial number, the resistance becomes elevated (29).
Takedown of the shunts during the period of rapid lung growth resulted in regression of both structural changes and pulmonary hypertension (30). Reversibility of medial hypertrophy
and muscularization of distal vessels has been shown in experimental studies following pressure off-loading (31). There are many common features linking the cellular and molecular pathophysiology of pulmonary vascular disease, regardless of etiology, to elevated elastase activity. This subject is discussed at the end of the chapter.
and muscularization of distal vessels has been shown in experimental studies following pressure off-loading (31). There are many common features linking the cellular and molecular pathophysiology of pulmonary vascular disease, regardless of etiology, to elevated elastase activity. This subject is discussed at the end of the chapter.
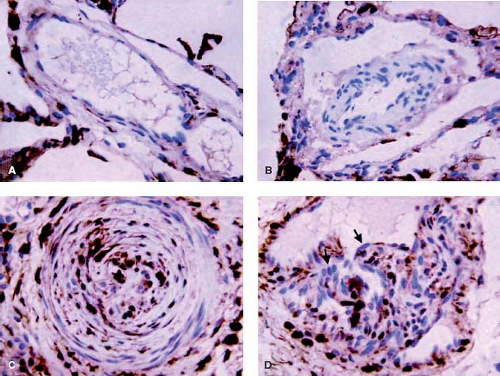 Figure 65.7 Representative photomicrographs of human lung biopsy tissue after immunoperoxidase staining for S100A4/Mts1. A: Vessel from patient graded 0-IB showing normal pulmonary artery with no immunodetectable S100A4/Mts1. B: Vessel showing a typical grade IB lesion with severe medial hypertrophy but without immunoreactivity for S100A4/Mts1. C: An artery from a patient with grade IVC disease with occlusive neointimal proliferation and strong positive staining for S100A4/Mts1 particularly in the intima compared to the media of the vessel. S100A4/Mts1 was not detected in all cells and appears to be localized in a subpopulation of intimal cells. D: A plexogenic lesion from a patient with grade IVC disease with staining of the smooth muscle cells and sparing of the endothelial cells (arrows). Immunoreactivity for S100A4/Mts1 was present in the lung parenchyma at a similar level in all grades of pulmonary vascular disease. Original magnification, ×40. (From Greenway S, van Suylen RJ, Du Marchie Sarvaas G, et al. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am J Pathol. 2004;164:253–262, with permission.) |
Heart Defects with Increased Pulmonary Venous Pressure
Vascular Changes
Although advanced pulmonary vascular disease is less likely to occur early in patients with congenital heart defects related to increased pulmonary venous pressure, Collins-Nakai et al. (32) reported greater than Heath–Edwards grade III changes in the majority of older children in their series with congenital mitral stenosis. In patients with total anomalous pulmonary venous connection, abnormal muscularization of the small arteries and veins was observed from birth, and more severe structural changes (i.e., Heath–Edwards grade III) were described in infants as young as 1 month of age. In infants with hypoplastic left heart syndrome, the early presence of severe medial hypertrophy of the small arteries and veins suggests that it developed in utero (33). In both adults and children with rheumatic mitral stenosis, Wagenvoort and Wagenvoort (34) observed increased medial wall thickness of the pulmonary arteries.
In postmortem arteriograms of infants and children with congenital heart defects and elevated pulmonary venous pressure, the axial pulmonary arteries have a reduced lumen diameter throughout their lengths. On microscopic examination of the lung, there is severe extension of muscle into peripheral intra-acinar arteries, which are normally nonmuscular, and failure of regression of the fetal musculature of the normally muscular arteries. The vessels, however, appear normal sized and are normal or slightly increased in number. The presence of pulmonary vascular changes in defects with high pulmonary venous pressure and the capacity for these abnormalities to regress with improvement in hemodynamics may be relevant because the Norwood and other various staged surgical procedures for the treatment of hypoplastic left heart syndrome ultimately depend on the success of a cavopulmonary shunt. Recent clinical experience suggests these patients may be quite symptomatic from pulmonary vascular disease and may respond well to cautious use of pulmonary vasodilator medications.
Experimental Studies
Pulmonary venous hypertension has been created experimentally by banding the pulmonary veins or by elevating left atrial pressure. LaBourene et al. (35) banded the pulmonary veins in newborn piglets and studied the pathophysiology of progressive pulmonary venous obstruction. After 1 week, they observed no change in pulmonary hemodynamics, but there was a striking decrease in compliance of the pulmonary veins. By 3 weeks, there was elevated pulmonary arterial pressure without a detectable elevation in pulmonary venous pressure. By 6 weeks, further progression of pulmonary hypertension was associated with elevated pulmonary venous pressure (Fig. 65.8). At 1 week, ultrastructural changes in the veins occurred that consisted of fragmentation of elastin and a phenotypic switch in medial smooth muscle cells in which they appeared to be more synthetic and migrating toward the subendothelium (Fig. 65.8). By 3 weeks, pulmonary venous hypertension developed and there was already evidence of intimal proliferation; by 6 weeks, there was an increase in collagen in the walls of the pulmonary veins. This study suggested that an increase in pulmonary arterial pressure may be the first sign of pulmonary venous obstruction and that elevated pulmonary venous pressure is associated with considerable structural remodeling.
Heart Defects with Decreased Pulmonary Blood Flow
Structural Features
In patients with congenital heart defects causing low pulmonary blood flow, hypoplasia of the pulmonary arterial musculature is observed, and in those in whom the hematocrit is particularly elevated, thromboemboli can occur. The creation of systemic-to-pulmonary artery shunts may improve the hypoplasia, but secondary to abnormally high flow and pressure, medial hypertrophy and intimal hyperplasia also could ensue. Studies suggest the potential for growth of the central pulmonary arteries is correlated with the proportion of elastin in the media, and the same may be true for the intrapulmonary vessels (36).
On postmortem arteriograms in patients with decreased pulmonary blood flow, the axial arteries have abnormally narrow lumen diameters (37). The background haze also may be reduced, as in patients with pulmonary atresia and intact ventricular septum. On microscopic examination of lungs from patients with pulmonary atresia and intact ventricular septum, the intra-acinar pulmonary arteries are abnormally thin-walled, small, and few; in patients with tetralogy of Fallot, these vessels are normal or decreased in muscularity, normal in number, and small (38). Alveolar development can be impaired as a consequence of decreased pulmonary blood flow, and this is reflected mostly by a reduction in alveolar number. Patients with tetralogy of Fallot and associated pulmonary atresia form a special subgroup in which the relative distribution of central pulmonary arteries and aortopulmonary collaterals determines peripheral pulmonary vascular structure. In patients with tricuspid atresia, the structural state of the pulmonary vascular bed is variable, depending on whether pulmonary blood flow is increased or decreased.
Experimental Studies
Levin et al. (39) created low pulmonary blood flow by banding the main pulmonary artery of fetal lambs. The resulting changes in the lung consisted of diffuse hypoplasia of the musculature of the peripheral pulmonary vessels; those identified were small in caliber and few in number. Haworth et al. (40) ligated the left pulmonary artery in newborn piglets and demonstrated normal development of the intra-acinar arteries owing to extensive anastomoses with bronchial arteries, but in the preacinar vessels, there was occlusive fibrosis of the lumen.
Hypoxia-Induced Pulmonary Vascular Disease
Hypoxia and Pulmonary Vasoconstriction
The pulmonary hypertension that occurs in response to acute hypoxia is usually mild and rapidly reversible. Hultgren et al. (41) demonstrated an 18% increase in pulmonary artery pressure in men brought suddenly from sea level to high altitude (7,800 ft). There is much variability in individual response to hypoxia; some people hyperventilate and become mildly alkalotic, hardly increasing their pulmonary artery pressure at all, whereas others develop severe pulmonary hypertension with high altitude pulmonary edema. Several theories have been proposed to explain high altitude pulmonary edema. Endothelial swelling of small arteries occurs in some areas of the lung and causes high resistance, which results in diversion of excessive flow through small vessels, causing edema. Defective fibrinolysis with formation of microemboli has been reported, as has inadequate diuresis (42). Abnormal endothelial metabolism of factor VIII has also been described. There is increased circulating antigenic activity without increased biologic activity, suggesting that the high–molecular-weight components of the molecule may
be associated with platelet microaggregates (43). Some studies suggest that high altitude pulmonary edema may have an immune basis because it is associated with major histocompatibility complex HLA-DR6 and HLA-DQ4; the subset with pulmonary hypertension is associated with HLA-DR6 (44). The combination of acetazolamide, NO and oxygen treatment has been successful, in alleviating the symptoms of high altitude pulmonary edema, and acetazolamide is also effective in preventing this complication.
be associated with platelet microaggregates (43). Some studies suggest that high altitude pulmonary edema may have an immune basis because it is associated with major histocompatibility complex HLA-DR6 and HLA-DQ4; the subset with pulmonary hypertension is associated with HLA-DR6 (44). The combination of acetazolamide, NO and oxygen treatment has been successful, in alleviating the symptoms of high altitude pulmonary edema, and acetazolamide is also effective in preventing this complication.
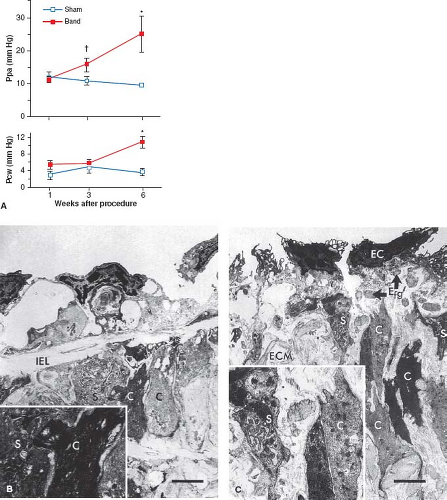 Figure 65.8 A: Pulmonary artery pressure (Ppa, upper panel) and pulmonary capillary wedge pressure (Pcw, lower panel) in banded and sham-operated piglets at 1, 3, and 6 weeks after banding. Values are averages of mean ± SEM (n = six piglets per group at each time point). At 1 week, there was no change. At 3 weeks, there was a significant increase in Ppa, which preceded the further rise in Ppa, at 6 weeks (*p <0.01; †p <0.05.). B, C: Transmission electron photomicrographs of representative pulmonary veins (PVs) from banded (C) and sham-operated (B) piglets at 3 weeks after banding (original magnification, ×14,131) (inset original magnification, ×28,263). Both pulmonary veins show apparent injury and lifting of endothelial cells and subendothelial spaces as a result of poor preservation during handling. Sham-operated PV displays on intact internal elastic lamina (IEL) and predominantly contractile-appearing smooth muscle cells (c) in media. In contrast, PV from banded piglet depicts complete breakdown of IEL into elastin fragments (Efg), a thickened subendothelium composed of collagen, extracellular matrix (ECM), and smooth muscle cells that appear to have migrated in from media, many of which have a synthetic-appearing phenotype (S) exemplified by a large amount of endoplasmic reticulum and a corresponding paucity of contractile filaments. C and S smooth muscle cells are better appreciated in insets. (Bar = 1 μm). (From LaBourene JI, Coles JG, Johnson DJ, Mehra A, Keeley FW, Rabinovitch M. Alterations in elastin and collagen related to the mechanism of progressive pulmonary venous obstruction in a piglet model. A hemodynamic, ultrastructural, and biochemical study. Circ Res. 1990;66:438–456, with permission.) |
Experimental Studies of Acute Hypoxia
Micropuncture has shown that both the small arteries and veins contribute to the acute hypoxic vasoconstrictor response. Studies by Hales et al. (45) suggested that prostaglandins mitigate the response to hypoxia, but the studies showed that the response to endothelial-derived relaxing factor is impaired. Recent reports revealed that with hypoxic vasoconstriction there is inhibition of a Ca2+-activated depolarizing potassium channel. With chronic hypoxia, this channel may become activated as a relaxing mechanism.
The relationship between acute hypoxic vasoconstriction and the sustained pulmonary hypertension and structural changes in the pulmonary vascular bed induced by chronic hypoxia is not known. Experimental studies of unilaterally banding the pulmonary artery in chronically hypoxic rats support the hypothesis that some structural changes in hypoxia are influenced by an alteration in the hemodynamics of the pulmonary circulation, and others are more direct effects of hypoxia per se (46). The direct effect of hypoxia is associated with increased ornithine decarboxylase activity, an enzyme implicated in structural remodeling of pulmonary arteries (47).
Chronic Hypoxia
Clinical Features of Chronic Hypoxia
In persons who live at high altitudes, there is chronic elevation in pulmonary artery pressure that is frequently reversible, at least under conditions of rest with administration of oxygen or return to sea level (48). In a study by Arias-Stella and Saldana (49), postmortem lung tissue from persons who had been living at high altitudes was compared with that obtained from sea-level dwellers. Structural changes were found in the vessels in the high altitude lung specimens; the peripheral arteries were more muscular than normal and had decreased lumen diameters.
Children living in Denver (elevation 5,200 ft) have slightly higher mean pulmonary artery pressures than sea-level dwellers, and children living in Morococha, Peru (elevation 14,900 ft) have mean pulmonary artery pressures that are twice as high. Moreover, in the high altitude residents, mean pulmonary artery pressure more than doubles with exercise, whereas it increases by only 50% in sea-level dwellers.
Experimental Studies
In animal studies, Tucker et al. (50) observed that the degree of reactivity to hypoxia or the level of pulmonary hypertension varied among different species according to the amount of smooth muscle in the pulmonary vascular bed. In the rat, the hemodynamic and structural responses of the pulmonary vascular bed to chronic hypoxia were studied. After just 3 days of chronic hypoxia, sustained elevations in pulmonary artery pressure and resistance were measured even after the rats were kept in room air for several hours. This finding coincides with the time that structural changes in the pulmonary vascular bed appear, in particular, the extension of muscle into peripheral arteries that are normally nonmuscular. Over the ensuing 2 weeks of hypoxia, mean pulmonary artery pressure progressively rises to double control values (51). This increase is accompanied by right ventricular hypertrophy, further extension of muscle into peripheral arteries, medial hypertrophy of normally muscular arteries, and reduction in arterial related to alveolar concentration. In the large preacinar pulmonary arteries, the adventitia is thickened as the result of an increase in the number of fibroblasts and collagen, and the media is also thicker as a result of hypertrophy of smooth muscle cells and accumulation of collagen, elastin, and other extracellular matrix components such as tenascin.
Relative hyporesponsiveness to hypoxia of the female animal has been observed in a number of species. During recovery from chronic hypoxia, mean pulmonary artery pressure returned to near normal in rats exposed as adults but remained 50% above normal in animals exposed during infancy, correlating with more severe residual vascular abnormalities. Ultrastructural and biochemical studies in the rat have shown that regression of smooth muscle hypertrophy following return to room air is accompanied by an increase in the amount of elastin and collagen in the vessel wall (52). Thus, the vessel, although less muscular, is remodeled in a way that may interfere with its compliance and its ability to grow. In studies by Kerr et al. (53), inhibition of collagen synthesis decreased chronic hypoxic pulmonary hypertension and vascular changes.
Stenmark et al. (54) took newborn calves to a simulated high altitude of 4,300 m and observed severe pulmonary hypertension with right-to-left shunting owing to the rapid development of suprasystemic levels of pulmonary artery pressure. There was striking medial hypertrophy and remarkable proliferation of a dense adventitial sheath, which, in large vessels, sometimes exhibited neovascularization (Fig. 65.9). Further studies showed striking synthesis of elastin in the pulmonary arteries of these neonatal calves. Most recent studies have implicated fibrocytes as key contributors to the remodeling of the pulmonary vasculature. It is believed that these “stem cells” that have characteristics of both fibroblasts and leukocytes (55) migrate into the vessel wall through the angiomata located in the expanding adventitia (56) (Fig. 65.10), and there is
also a population of stem cells in the adventitia that may expand under conditions of hypoxia (57). Fibroblasts may also play a role in regulation of inflammation and remodeling in the pulmonary vasculature. Recently, in bovine and rat models of hypoxia-induced pulmonary hypertension and in patient tissue, adventitial fibroblasts from hypertensive arteries were shown to activate macrophages through paracrine IL-6, STAT3, HIF1α, and C/EBPβ signaling to drive expression of genes involved in inflammation and remodeling (58). There is also evidence in the pulmonary hypertensive calf model that epigenetic factors, specifically a decreased level of microRNA 124, may be controlling the expression of proinflammatory cytokines in activated fibroblasts and that the inflammatory response is critical to the evolution of the disease and may be controlled by histone deacetylase inihibition (59,60).
also a population of stem cells in the adventitia that may expand under conditions of hypoxia (57). Fibroblasts may also play a role in regulation of inflammation and remodeling in the pulmonary vasculature. Recently, in bovine and rat models of hypoxia-induced pulmonary hypertension and in patient tissue, adventitial fibroblasts from hypertensive arteries were shown to activate macrophages through paracrine IL-6, STAT3, HIF1α, and C/EBPβ signaling to drive expression of genes involved in inflammation and remodeling (58). There is also evidence in the pulmonary hypertensive calf model that epigenetic factors, specifically a decreased level of microRNA 124, may be controlling the expression of proinflammatory cytokines in activated fibroblasts and that the inflammatory response is critical to the evolution of the disease and may be controlled by histone deacetylase inihibition (59,60).
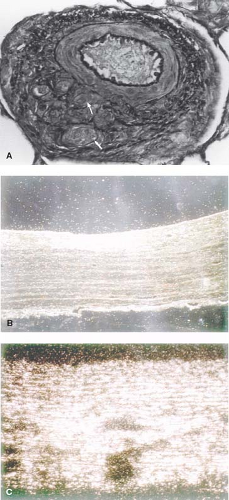 Figure 65.9 An artery from the lung of a 2-week-old calf raised at a simulated altitude of 4,300 m from birth. Pulmonary artery systolic pressure was 100 mm Hg. A: There is marked medial hypertrophy and adventitial thickening with neovascularization (arrows). Elastic tissue stain, original magnification, ×400. (Kindly supplied by K. Stenmark.). In situ hybridization localization of tropoelastin mRNA in control and hypertensive vessels from neonatal calves. White staining over areas indicates tropoelastin mRNA labeling. In normotensive vessels (B), labeled cells (35S-labeled T66-T7) were confined to the inner media. Minimal signal is noted in the outer vessel wall. In vessels from hypertensive animals (14 days of hypoxia) (C), intense autoradiographic signal was observed throughout the media, albeit in a patchy distribution. (From Prosser IW, Stenmark KR, Suthar M, Crouch EC, Mecham RP, Parks WC. Regional heterogeneity of elastin and collagen gene expression in intralobar arteries in response to hypoxic pulmonary hypertension as demonstrated by in situ hybridization. Am J Pathol. 1989;135:1073–1088, with permission.) |
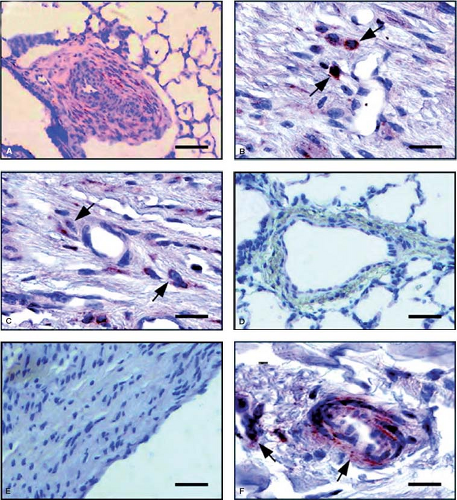 Figure 65.10 Immunohistochemistry (brown peroxidase signal) revealed a greater number of c-kit+ cells (arrows) in the vessel wall of distal (A) and proximal (B, C) arteries from control animals. The c-kit+ cells (arrows) are localized contiguous to vasa vasorum in proximal vessels (B, C). Scale bar represents 100 μm in A, D, and E, and 50 μm in B, C, and F. (From Davie NJ, Crossno JT Jr, Frid MG, et al. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L668–L678, with permission.) |
Studies in transgenic mice suggest that genetic factors might modulate the response to chronic hypoxia. For example, in the absence of heme oxygenase 1, there is reduced production of NO and its associated vasodilatory effects (61). Prostacyclin synthetase overexpression is protective against the hemodynamic and vascular changes of pulmonary hypertension (62). Serotonin has been implicated in the increased vasoreactivity of the Fawn-hooded rat (63), and there is attenuated severity of pulmonary vascular disease in mice lacking the SERT gene (64). Overexpression of SERT worsens hypoxia-induced pulmonary hypertension (65) as does haploinsufficiency of bone morphogenetic protein receptor 2 (BMPR2) (66) or dominant negative BMPR2 (67), although in both cases the degree of structural remodeling is relatively unimpressive. Haploinsufficiency of BMPR2 in cultured smooth muscle cells and in transgenic mice increases their sensitivity to the pro-proliferative effects of serotonin (68). Epigenetic factors also appear to control signaling via BMPR2 (69) as well as reduced expression of the free radical scavenger superoxide dismutase seen in the Fawn-hooded rat with pulmonary hypertension (70).
We have shown that mice overexpressing the calcium binding protein S100A4/Mts1 have mild pulmonary hypertension under room air conditions. Values are increased over control mice in hypoxia but the remodeling response also appears to be mitigated. We related this to increased production of fibulin-5 and thickening of the elastic laminae (71). Thus overexpression of genes that might worsen hypoxia-induced pulmonary hypertension appears to invoke compensatory mechanisms that protect against the remodeling response. Understanding why these compensatory mechanisms fail may be critical in appreciating why some patients develop rapidly progressive pulmonary hypertension.
Numerous studies have attempted to show how acute vasoconstriction or a direct hypoxic “injury” initiates the structural changes observed in the pulmonary arteries. There is convincing evidence that the high endothelin levels are causally related to hypoxic vasoconstriction and the subsequent initiation of vascular changes. In some studies, chronic hypoxia appears to decrease endothelial nitric oxide synthase (eNOS). Strategies proven effective in the treatment of chronic hypoxic pulmonary hypertension in rats include inhibition of 5-lipoxygenase activating protein (FLAP) as well as inhibition of cyclic 3′-5′-guanosine monophosphate-specific phosphodiesterase, and continuous infusion of NO. In other studies, activation of voltage-gated K channels (Kv) by gene transfer or a metabolic activator inhibited chronic hypoxic pulmonary hypertension (72,73). Serine elastase inhibitors also effectively reduce chronic hypoxia-induced pulmonary hypertension and associated vascular remodeling (74). In addition, experimental studies suggest that vascular smooth muscle growth inhibitors may be useful in preventing vascular disease. Heparin infusion will decrease the severity of hypoxia-induced vascular changes, presumably by decreasing smooth muscle hyperplasia. Most recently rho kinase inhibitors have proven effective, when administered even by inhalation, in preventing pulmonary hypertension and structural changes associated with chronic hypoxia (75,76,77,78).
Lung Disease
Several factors may contribute to the pulmonary hypertension that commonly but not invariably accompanies severe parenchymal lung disease: the level of hypoxia and polycythemia, the degree of endothelial injury and subsequent imbalance in vasoactive mediators and vascular reactivity, the level of pulmonary venous pressure secondary to dysfunction of the hypoxemic left ventricle, and the nature and severity of the structural damage to the pulmonary arteries. Treatment of the lung disease will decrease the level of pulmonary artery pressure by eliminating the contributing causes and by allowing some regression of the structural changes. The latter, however, ultimately determines the level of pulmonary hypertension and the rate of development of right-sided heart failure (cor pulmonale).
Obstructive Lung Disease
In children, unlike in the adult population, obstructive lung disorders such as asthma are only rarely associated with the development of pulmonary hypertension. In patients with cystic fibrosis, however, cor pulmonale is more common. When right ventricular hypertrophy and failure are observed, they generally reflect the degree of abnormal structural remodeling of the pulmonary vascular bed. The severity of the arterial changes tends to be patchy, reflecting the nature of the lung disease, but the venous changes are more uniform, suggesting that they result from left atrial hypertension secondary to the left ventricular dysfunction.
Restrictive lung disease may occur in childhood from a variety of causes, and pulmonary hypertension is a common complication. Examples of such restrictive lung disorders are diffuse interstitial fibrosis (Hamman–Rich syndrome), bronchopulmonary dysplasia, radiation fibrosis and chemotherapy toxicity, infiltrative lung tumors, and collagen vascular disease. In the latter, pulmonary vasculitis may be important etiologically and is discussed separately. Reversal of the pulmonary hypertension generally depends on the ability to affect the course of the interstitial lung disease. Few structural studies have been done of patients with restrictive lung disorders. It has been proposed that, secondary to fibrosis of the alveolar septa, the loss of small alveolar wall arteries results in an increase in pulmonary vascular resistance, and as a secondary response there is hypertrophy of normally muscular arteries. In a study of the lungs from premature infants in whom bronchopulmonary dysplasia was a complication of severe respiratory distress syndrome, Rendas et al. (79) observed that, although the preacinar arteries had dilated appropriately, there was extension of muscle into normally nonmuscular peripheral arteries, which might explain the persistent pulmonary hypertension in these patients. In addition, although the number of peripheral arteries relative to alveoli was normal, the marked reduction in alveolar number suggested an absolute decrease in the cross-sectional area of the pulmonary vascular bed. As the clinical definition of bronchopulmonary dysplasia evolves from the fibroproliferative process described by Northway et al. (80) to the “new” bronchopulmonary dysplasia associated with extreme prematurity and reduced pulmonary vascular growth, there is a weaker correlation between severity of lung disease and pulmonary hypertension, suggesting the pulmonary hypertension may more directly related to compromised vascular development (81).
Upper Airway Obstruction
Severe upper airway obstruction from a variety of causes (Fig. 65.11 and Table 65.5) may be complicated by the development of pulmonary hypertension. In each case, the hypertension is not always a direct result of the degree of airway obstruction but seems to depend also on whether there is a central component, such as diminished ventilatory drive or general neuromuscular pharyngeal dysfunction.
Many patients with upper airway obstruction have worsening of their symptoms with sleep; this worsening may be mechanical, such as positioning of the head, or it may be the result of a central mechanism. Although removal of the airway obstruction often results in prompt return to normal pulmonary artery pressure and resolution of heart failure, often these symptoms persist for some time perhaps because of slow regression of hypoxia-induced structural changes in the pulmonary vascular bed or persistent impairment of ventilatory drive.
Many patients with upper airway obstruction have worsening of their symptoms with sleep; this worsening may be mechanical, such as positioning of the head, or it may be the result of a central mechanism. Although removal of the airway obstruction often results in prompt return to normal pulmonary artery pressure and resolution of heart failure, often these symptoms persist for some time perhaps because of slow regression of hypoxia-induced structural changes in the pulmonary vascular bed or persistent impairment of ventilatory drive.
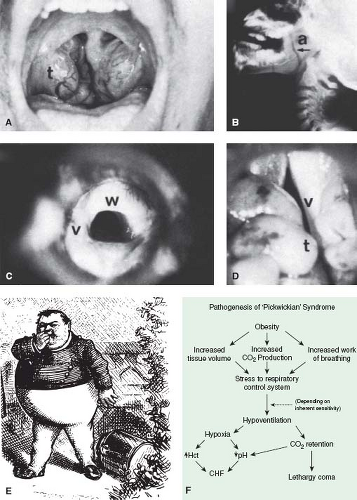 Figure 65.11 A–D: Four different causes of upper airway obstruction. A: Hypertrophied tonsils (t). B: X-ray of head and neck demonstrating compression of nasopharynx (arrow) by hypertrophied adenoids (a). C: Glottic web (w) adjacent to vocal cord (v). D: Lingual tonsils (t). E: Thomas Nast’s drawing of the fat boy in “The Pickwick Papers” from an American edition of the Posthumous Papers of the Pickwick Club, London, 1837; New York, 1873. F: A schema of the pathogenesis of the pulmonary hypertension in the Pickwickian syndrome. (Modified from Auchincloss JH Jr, Gilbert R. The cardiorespiratory syndrome related to obesity: clinical manifestations and pathologic physiology. Prog Cardiovasc Dis. 1959;1:423–434.) |
TABLE 65.5 Causes of Upper Airway Obstruction in Children | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
The Pickwickian syndrome described in adults, consisting of hypersomnolence and obesity associated with pulmonary hypertension, also has been reported in children (Table 65.5). The obesity, through increased work of breathing, stresses the respiratory control system, and depending on its inherent sensitivity, hypoventilation may result, causing further retention and hypoxia, which contribute to the lethargy and cor pulmonale (Fig. 65.11). Mental retardation is often but not invariably associated with this syndrome. Damage to the respiratory center, either as a primary disorder (Ondine curse) or secondary to trauma or other neurologic disease, also may result in cor pulmonale as a result of chronic intermittent hypoxia and hypercarbia. Although the pulmonary
hypertension of sleep apnea in adults is in large part attributable to airway obstruction, in children, it is often the manifestation of an abnormal central mechanism. Increasing attention to the “metabolic syndrome” and systemic vascular disease has prompted us to investigate insulin resistance which occurs with obesity and is also related to the development or progression of pulmonary hypertension (82).
hypertension of sleep apnea in adults is in large part attributable to airway obstruction, in children, it is often the manifestation of an abnormal central mechanism. Increasing attention to the “metabolic syndrome” and systemic vascular disease has prompted us to investigate insulin resistance which occurs with obesity and is also related to the development or progression of pulmonary hypertension (82).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


