Chapter 37 Pathophysiology, Epidemiology, and Prognosis of Aortic Aneurysms
Aortic aneurysms result in significant morbidity and mortality, accounting for nearly 13,000 deaths and 55,000 hospital discharges per year in the United States.1 Although aneurysms may affect any part of the aorta from the aortic root down to the abdominal aorta, the prognosis and outcome in patients with aortic aneurysms vary based on location and underlying etiology. Timely and appropriate intervention may improve the natural history of the disease process. This chapter reviews the pathophysiology, epidemiology, and prognosis of aortic aneurysms.
The Normal Aorta
The aorta is the large conduit vessel through which the heart delivers blood to the entire body. It courses from the heart through the thorax and abdomen, and ultimately bifurcates into the common iliac arteries (CIAs) in the abdomen. In the thorax, the aorta can be subdivided into three segments: ascending aorta (from the base of the heart to the innominate artery), transverse aorta or aortic arch (including the great vessels and extending to the left subclavian artery), and descending aorta (from the distal edge of the subclavian artery to the level of the diaphragm) (Fig. 37-1).
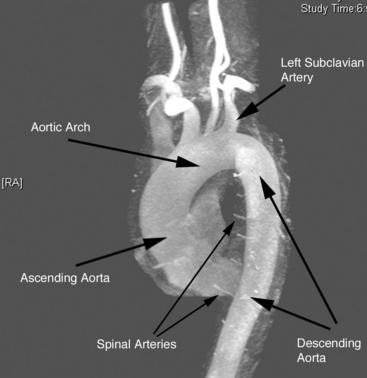
Like other arterial structures, the aorta is composed of three layers: tunica intima, tunica media, and adventitia. The innermost surface of the tunica intima is lined by a single-cell-thick layer of endothelial cells (ECs). The intima is bound by the internal elastic lamina. The tunica media is composed of smooth muscle cells (SMCs), collagen, fibroblasts, elastin fibers, and ground substance, which together control the degree of vessel constriction and vasodilation. The presence of elastin fibers in the media defines the aorta as an elastic artery and provides the tensile strength that permits the aorta to withstand pulsatile delivery of blood from the heart. Elastin content gradually decreases with distance from the heart.2 The outermost layer, the adventitia, is a thin layer that contains connective tissue, fibroblasts, and the nutritive vasa vasorum.
Definition of Aortic Aneurysm
In adults, the normal diameter of the aorta is approximately 3 cm at the origin, 2.5 cm in the descending thoracic aorta, and 1.8 to 2 cm in the abdominal aorta. Aortic aneurysm is defined as a maximal aortic dimension greater than 3.0 cm, or a 50% increase in size compared with the normal segment proximal to the aneurysm. Mild expansion that does not meet these criteria may be referred to as aortic ectasia. True aneurysms are classified into two major groups on the basis of morphology: (1) fusiform (Figs. 37-2 and 37-3), defined as a circumferential expansion of the aorta, and (2) saccular, representing a focal outpouching of a segment of the aorta (Fig. 37-4). Fusiform aneurysms are the most common manifestation. In contrast to true aneurysms, which involve expansion of all three layers of the aortic wall, a pseudoaneurysm, also known as a false aneurysm, results from a disruption of the aortic wall and essentially represents a contained rupture of the aorta.
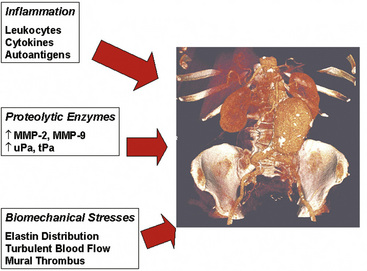
Figure 37-2 Three pathophysiological mechanisms that best characterize the process of aneurysm formation.
(Volume-rendered computed tomography image of abdominal aortic aneurysm used with permission of Joseph Schoepf, MD.)
Pathophysiology of Aortic Aneurysms
A wide variety of pathological states are associated with aortic aneurysms (Box 37-1). These include degenerative diseases, inherited disorders, infections, inflammatory conditions (i.e., vasculitis), and trauma. Specific disorders associated with aortic aneurysms are discussed later in this chapter. Important determinants of aortic aneurysm formation include inflammation, proteolysis of the structural components of the aortic wall, and abnormal biomechanical forces3 (see Fig. 37-2). Understanding the underlying pathophysiology of aneurysm formation is critical not only for prevention of initial aneurysm formation but also for limiting aneurysm growth and expansion.
 Box 37-1
Box 37-1Traditionally, pathological aortic aneurysm formation was ascribed to a process akin to atherogenesis. Although advances in basic and clinical investigation in both lesion types have revealed some common themes, newer studies suggest that aneurysm formation is fundamentally different from atherosclerosis. Preferential weakening of the adventitia and media—rather than an intimal proliferative process, as in atherosclerosis—results in diminished aortic resilience and tensile strength, culminating in aortic wall thinning, dilation, and increased wall stress, all of which may result in rupture. Although atherosclerotic changes may be seen in the wall of aneurysms, these changes may be a consequence of local turbulent flow as opposed to a cause of aneurysm formation.4,5 Moreover, the degree of systemic atherosclerosis does not correlate well with the degree of aneurysm formation.6
Development of aneurysms is associated with loss of two critical structural elements in the aortic wall: elastin and collagen. Elastin provides radial and longitudinal support, enabling the aorta to respond to pulsatile flow while maintaining normal arterial dimensions. The importance of elastin in maintaining aortic structure is highlighted by animal models where elastase infusion results in elastin breakdown and experimental aortic aneurysm formation.7 However, breakdown of elastin alone appears insufficient to cause aneurysmal expansion and rupture. Loss of collagen, another important structural element, is an additional contributor, and the relative balance of elastin and collagen deposition, among other factors, may be critical for determining aneurysm formation.8 Early in aneurysm formation, the aorta compensates for loss of elastin by increasing production of collagen,8 but as elastin content decreases, collagen (as the major source of tensile strength) is overwhelmed, and aortic expansion occurs. This is exacerbated by up-regulation of collagenases, resulting in further collagen degradation as described later.9 Structural changes in each layer of the aortic wall develop that together promote aortic stiffness. As a consequence, decrease in the vessel’s ability to distend normally with left ventricular (LV) contraction, weakening of the vessel wall, and increase in the tendency for dilation and ectasia follow.10 Some of the changes in aortic structure that promote aneurysm formation may arise as a result of the normal aging process. With normal aging, aortic stiffness due to fragmentation of elastin fibers, deposition of glycosaminoglycans, fibronectin (FN), and collagen, and reduced bioavailability of endothelium-derived nitric oxide (NO) occurs.11–14
Pathophysiologically, the major determinants of aortic aneurysm formation include proteolysis of the structural components of the aortic wall, inflammation, and abnormal biomechanical forces3 (see Fig. 37-2). Pathology of aortic aneurysms varies in different segments of the aorta and in different predisposing diseases. Frequently observed histological features include cystic medial necrosis, mucoid infiltration, and cyst formation in the setting of elastin necrosis and vascular smooth muscle apoptosis. In patients with Marfan’s syndrome (MFS), bicuspid aortic valve (BAV), or Turner’s syndrome, cystic medial necrosis is a common feature, but in contrast, inflammation is less prominent than in abdominal aortic aneurysms (AAAs).15,16 On the other hand, cystic medial necrosis is less likely to be observed in AAAs. Instead, AAAs typically have disrupted elastin fibers, inflammation, greater vascular smooth muscle cell (VSMC) apoptosis, and deficient glycosaminoglycan production.17 Elastin fragmentation occurs adjacent to the inflammatory cells. Despite differences in pathophysiology due to location and underlying etiology, formation of all aortic aneurysms involves to some degree the processes described in the following discussions (i.e., proteolytic degradation, inflammation, changes in biomechanical forces) that together facilitate aneurysm formation.
Proteolytic Degradation
Several proteolytic enzymes contribute to degradation of structural components of the arterial wall, ultimately increasing risk of aneurysm formation. Matrix metalloproteinases (MMPs) are endopeptidases that degrade one or more components of the extracellular matrix (ECM). Thus far, several classes of MMPs, comprising nearly 30 individual proteinases, have been characterized. They include collagenases, gelatinases, stromelysins, matrilysins, membrane-type (MT)-MMPs, and other MMPs.18 Typically produced as proenzymes, MMPs may be activated both intracellularly and extracellularly, and may be secreted by endothelial cells, vascular smooth muscle cells, or adventitial fibroblasts. Extracellular regulation of activation may occur as a result of an MMP–MT-MMP interaction or via interaction with plasmin or reactive oxygen species (ROS).19–21 The inhibitors of MMPs are the tissue inhibitors of matrix metalloproteinases (TIMPs) and plasminogen activator inhibitors (PAI) 1 and 2.18
Increased local production of MMPs in aortic aneurysms was first reported nearly 20 years ago.22–24 Although elevations of several MMPs have been noted, MMP-1 (collagenase) and MMP-3 (stromelysin), MMP-2 (gelatinase A), and MMP-9 (gelatinase B) represent the principal proteinases in aortic aneurysms that result in elastin and collagen degradation.25,26 MMP-2 and MMP-9 gelatinases specifically break down collagen. They are synthesized by local cells in the aortic wall, including infiltrating macrophages and resident aortic VSMCs.27,28 MMP-9 is typically found in the adventitia near the vasa vasorum, localizing to infiltrating macrophages.29 MMP-2 is synthesized constitutively by VSMCs30 but can also be synthesized by infiltrating leukocytes.31 Interestingly, MMP-2 production is increased in the vasculature remote from the aorta, suggesting a systemic underlying disease process that manifests with aortic aneurysmal disease.32
The centrality of these enzymes in aneurysm formation is supported by several lines of evidence. Studies have demonstrated that MMP-2 and MMP-9 levels are higher in tissue obtained from aortic aneurysms than in atherosclerotic plaque or normal arterial tissue.30,31 Furthermore, MMP-9 and MMP-2 knockout mice do not form aortic aneurysms in experimental models.33 Reinfusion of competent macrophages from wild-type mice into MMP-9 knockout mice enables aneurysm formation.33 In addition, elevated levels of MMP-1, MMP-8 (a neutrophil collagenase), and MMP-9 have been associated with aneurysm rupture, and levels of these MMPs may vary with aneurysm size.34–36 Relationship to aneurysm size is less clear for MMP-2.37–40
Elevated levels of other proteolytic enzymes such as human macrophage metalloelastase (MMP-12) and membrane type-1 metalloproteinase (MT1-MMP) have also been demonstrated in aortic aneurysms.41 Expression of MMP-12 is increased in aortic aneurysms as a result of macrophage infiltration. However, the relevance of MMP-12 to aneurysm formation is less clear, based on the finding that MMP-12 knockout mice are not completely protected from aneurysm formation.42 MT1-MMP, a collagenase produced by macrophages and increased in aortic aneurysms, likely has its greatest effect as an activator of the proenzyme form of MMP-2.43,44
In addition to increased expression of MMPs, aneurysm formation is associated with abnormal regulation of MMP levels in tissues. Matrix metalloproteinase levels are increased in states of inflammation and oxidant stress, both known to play a role in aneurysm formation. Compared with nonaneurysmal sections of aorta, superoxide anion and markers of oxidative stress are increased in aneurysmal segments, and ROS can convert proenzymes of MMPs to their active form.45–47 In addition, MMP levels can be augmented by plasminogen activators, such as urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), which are specific physiological regulators of MMP-2 and MMP-9 activation and overexpressed in aortic aneurysms but not in healthy aortic specimens.48
Matrix metalloproteinase levels are also normally regulated by TIMPs. Although early work reported decreased concentrations of TIMPs in aneurysmal tissue,49 more recent data have been less clear, showing no difference in certain TIMP levels between aneurysmal and healthy aortic tissue.36,43,50,51 However, the relative imbalance in MMPs to TIMPs may be the more relevant factor.41 Nonetheless, experimental data have clearly demonstrated the significance of TIMPs to aneurysm formation. Local overexpression of TIMP-1 prevented aneurysm formation in a rat model of aortic aneurysm,52 but TIMP knockout mice have increased MMP activity and greater aneurysm formation.53 Similarly, lower expression of PAI-1, a regulator of plasminogen activator activity, has been reported in aneurysmal disease.49,54 Increased expression of plasminogen activators without reciprocal changes in their inhibitors alters the balance toward fibrinolysis, MMP activation, and tissue degradation. Experimental overexpression of PAI-1 prevents aneurysm formation in a rat model of aortic aneurysm,55 confirming the importance of relative rather than absolute concentrations.
The contribution of MMPs to the pathophysiology of aortic aneurysms is highlighted by the beneficial effect of medications known to reduce MMP levels on aneurysm expansion. For example, tetracyclines (e.g., doxycycline) have long been recognized as generalized inhibitors of metalloproteinases.56 Their potential value in aortic aneurysmal disease has been demonstrated in both organ culture and rodent models of aneurysm formation.36,57–59 In humans, doxycycline limits the activity of both MMP-2 and MMP-9 in aortic wall specimens, both by reducing macrophage MMP-9 messenger ribonucleic acid (mRNA) expression and diminishing activation of the proenzyme form of MMP-2.60–62 In a few small randomized phase 2 trials, doxycycline has been shown to decrease aneurysm expansion rate.63,64 Although these data await confirmation in larger trials, they do support the conceptual framework of the importance of proteolytic enzymes in aortic aneurysm formation and expansion.
By reducing levels of proteolytic enzymes, several other therapies may potentially be of therapeutic benefit in aneurysmal disease. HMG-CoA reductase inhibitors, or statins, have antianeurysm properties in experimental models by virtue of reducing oxidant stress and macrophage production of MMPs.65–67 However, data in humans have been inconsistent. A meta-analysis of five studies including 697 patients with small aortic aneurysms (<55 mm) treated with or without statins suggested that statin therapy was associated with lower rates of expansion.68 On the other hand, other human studies have not shown a benefit of preoperative statin treatment on MMP or TIMP levels in aneurysm specimens, and no difference in aneurysm expansion rate.69–71 Indomethacin also appears to prevent aortic aneurysm formation in a rat model,72 and the mechanism may be related to inhibition of cyclooxygenase (COX) 2, prostaglandin E2 (PGE2), and reductions in MMP-9.73–75 Indeed, PGE2 expression is up-regulated more than 30-fold in aortic aneurysms.76 Prostaglandin E2 localizes to infiltrating macrophages, and its expression is dependent on COX activity.77 Prostaglandin E2, through activation of interleukin (IL)-6, may increase VSMC apoptosis, further weakening the structural elements of the aorta.75,76,78
Inflammation
The contribution of inflammation to many pathological arterial processes has been well established.79 In 1981, Rose and Dent80 reported mild chronic inflammation in 72.5% and moderate inflammation in 15.7% in 51 consecutively resected AAAs. Subsequently it was discovered that lymphocytes and macrophages are found in greater quantity in the adventitia and media of AAAs than of atherosclerotic or normal aortas.81 In addition, surgical explant specimens from patients with aortic aneurysms demonstrated higher levels of adhesion molecules, including intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1, than seen in atherosclerotic and normal aortas. Similarly, tissue levels of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-8, monocyte chemoattractant protein (MCP)-1, interferon (IFN)-γ, and IL-6, have all been noted to be elevated in patients with aneurysms compared with control subjects.82 Some studies have even suggested cytokine levels are higher in ruptured aortic aneurysms than in asymptomatic aortic aneurysms,83 although the data have been inconsistent.84 Finally, other acute-phase proteins, including C-reactive protein (CRP), D-dimer, and ceruloplasmin, are also present at increased levels in plasma and in the vessel wall.82,85–87
These inflammatory mediators largely derive from infiltrating macrophages, but lymphocytes and aortic ECs and SMCs also contribute to the inflammatory milieu. The presence of inflammation in aortic aneurysms is supported by positron emission tomographic imaging with18 fluorodeoxyglucose (FDG-PET) showing greater FDG uptake in aneurysmal compared to nonaneurysmal aortic segments from matched control subjects, and by advanced magnetic resonance imaging (MRI) techniques.88,89 Thus, immune cells invade the aortic wall, become activated, and create an inflammatory environment that engages the activity of local stabilizing cells, initiating the process of elastin and collagen breakdown and aneurysm formation. However, the initial signals that drive inflammatory cell recruitment remain unclear. Animal studies with experimental aneurysm models have confirmed the human studies and demonstrated that increased inflammation promotes aneurysm formation.90
Given the contribution of inflammation to aortic aneurysms, it follows that strategies to reduce inflammation might reduce aneurysm formation or limit aneurysm growth. Indeed, as mentioned previously, some data suggest that statins, known to have beneficial antiinflammatory properties beyond their effect on cholesterol lowering, may limit aneurysm growth and expansion.67,91,92 Recent studies have also shown that limiting inflammation can reduce aneurysm formation in animal models.93–97 Future studies will be required to clarify whether strategies to target inflammation can prevent formation of aortic aneurysms or limit expansion in humans.
Increases in Biomechanical Wall Stress
Several specific structural changes may predispose the abdominal aorta to aneurysm formation. For example, elastin within the aortic wall is organized into circumferential plates, or lamellae, that respond to the pulsatile load created by the heart. Each lamellar unit consists predominantly of two elastin bundles and vascular smooth muscle. However, deposition of elastin is not uniform along the aorta, with the thoracic aorta incorporating 35 to 56 lamellar units compared to only 28 in the abdominal aorta.98 The abdominal aorta may therefore be more susceptible to elastin breakdown due to a relative increase in pressure withstood per lamellar unit, compared with the rest of the vessel. In addition, the abdominal aorta has a decreased concentration of nutritive vasa vasorum compared to more proximal aortic segments.99 Reductions in aortic tissue perfusion stiffen the vessel, reducing compliance and ability to withstand pulsatile stress.100
Vascular cells in the aorta attempt to restore elastin content in the setting of elastin degradation to compensate for reductions in tensile and radial strength. Human AAA samples show a four- to sixfold increase in tropoelastin protein compared with control arteries.101,126 Elastin is produced by SMCs and macrophages. In areas of macrophage infiltration, elastin deposition is not organized into mature effective bundles. Indeed, compared with normal specimens, aneurysm specimens have a ninefold reduction in desmosine, a marker for mature elastin cross-linking.101 Thus, compensatory elastin replacement is disordered and does not improve aortic compliance.
Another factor that may make the abdominal segment of the aorta more prone to aneurysm formation is blood flow patterns specific to that segment. In experimental models, the infrarenal segment of the aorta is subject to much higher levels of oscillating flow and reflected pressure waves compared with the suprarenal segment,102 resulting in higher levels of aortic wall tension. Turbulence and pressure are exacerbated by the aneurysm’s morphology, which promotes development of local vortices and turbulent flow patterns.103 Excluding these flow patterns from the aneurysm by placing an aortic endograft rapidly reduces plasma MMP-9 levels in patients.104,105 In a rodent elastase infusion model of aortic aneurysm, flow conditions were examined by creating a left femoral arteriovenous fistula (AVF) or left iliac artery ligation. Increases in shear stress due to fistula formation resulted in a more stable aortic phenotype with decreased oxidative stress, decreased macrophage density in the media, increased aortic ECs and vascular smooth muscle cells, and reduced apoptosis compared to the lower shear stress introduced by femoral artery ligation.4,106,107 Improved flow decreased aortic expansion by 26%.106
In addition to adverse biomechanical forces creating an environment permissive for aneurysm formation, the role of intraluminal thrombus on wall stress and aneurysm expansion has recently been a focus of much study. Using finite element analysis in a three-dimensional (3D) model of the aorta derived from computed tomography (CT) scans, intraluminal thrombus was found to lower peak wall stress by up to 38%108 relatively independent of thrombus constituents.109 Thrombus decreases transmission of luminal pressure to the aneurysm wall and may prevent aneurysm rupture by reducing wall strain.110–115 On the other hand, intraluminal thrombus may also contribute to further aneurysm formation,111 given that thrombus has been demonstrated to have higher levels of proteolytic enzymes and enzyme activators than the aneurysm itself.116 Intraluminal thrombus may act as a proteolytic enzyme reservoir through polymorphonuclear leukocytes, and enzyme accumulation provides a ready source of destructive elements for the adjacent aneurysm.116
Understanding biomechanical factors may help improve assessment of risk of aneurysm growth and rupture above and beyond the predictive value of lumen diameter alone.117 Several studies have used novel computational models to incorporate biomechanical factors such as wall stress, wall strength, and extent of intraluminal thrombus for assessment of abdominal aneurysms.112,118–123 Some studies have suggested that assessment of wall stress, wall strength, and the ratio of wall stress to wall strength may be better predictors of rupture risk than diameter.124,125
Epidemiology and Prognosis of Aortic Aneurysms
Abdominal Aortic Aneurysms
Prevalence
The prevalence of aneurysms of the abdominal aorta has been determined on the basis of several large screening studies and autopsy series (Table 37-1). In an early series of 24,000 consecutive autopsies performed over 23 years, 1.97% of the subjects were found to have an AAA.126 Of the 473 aneurysms found, 58% were larger than 4 cm in diameter, nearly three quarters of the patients were men, and one fourth of the aneurysms had ruptured. More recent large screening programs in targeted populations have further evaluated the prevalence of AAA. The largest screening program performed was the Aneurysm Detection and Management (ADAM) Study Screening Program, which studied 126,196 veterans 50 to 79 years of age.127 In this cohort of predominantly male American veterans, 3.6% of subjects had an infrarenal aortic diameter greater than 3 cm, and an AAA 4 cm or larger was found in 1.2%. The Multicentre Aneurysm Screening Study (MASS) also screened 27,147 of 33,830 invited men aged 65 to 74 and reported a 4.9% prevalence of AAA 3 cm or larger.128
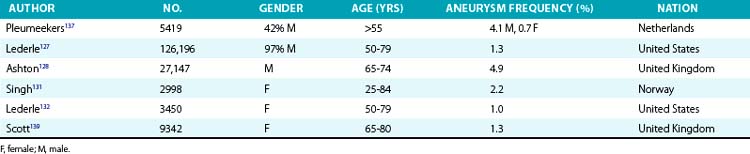
Several studies have demonstrated lower prevalence of AAA in women. The largest and most recent of these studies screened nearly 10,012 women (mean age, 69.6 years) and found an AAA prevalence rate of 0.7%, with only 4 of 74 detected aneurysms measuring larger than 5 cm.129 These low prevalence rates were consistent with findings from earlier studies. Among 4237 subjects aged 65 to 80 who participated in a screening study among general practitioners in West Sussex, United Kingdom, 2290 women agreed to undergo abdominal ultrasonography.130 Only 1.4% of the women had an AAA 3 cm or larger, and only 0.3% had an AAA 4 cm or larger. This dramatically lower prevalence in women has been confirmed in subsequent studies. In the Norwegian Tromso study, 2% of 2943 women aged 55 to 84 had an AAA 3 cm or larger, and 0.5% had an abdominal aneurysm 4 cm or larger.131 Finally, among female American veterans, the prevalence of an AAA 3 cm or larger was just 1%.132 However, an increasing number of cardiovascular risk factors does increase the risk of AAA in women, with a prevalence rate as high as 6.4% in this higher-risk group.129
Risk factors
Three risk factors predict the vast majority of AAAs: age, gender, and cigarette smoking. Aneurysms usually affect the elderly, seldom occurring in those younger than 60 years of age, and there is a clear increase in incidence with increasing age, even when limiting the studies to older individuals.133–135 In a Norwegian population-based study of 6386 men and women aged 25 to 84, the incidence of AAA in men increased from 0% in those aged 25 to 44, to 6% in those aged 55 to 64, and to 18.5% in those aged 75 to 84.131 Large North American epidemiological studies have demonstrated an increase in AAA risk ranging from 58% to 300% with each additional decade of life.136 Gender is also an important predictor of AAA formation; in all age groups, risk of AAA is two- to sixfold higher in men than women.131,132,137–140 Cigarette smoking is the most potent modifiable risk factor and increases the risk of AAA by 60% to 850%.141–147 In the ADAM study and the Edinburgh Artery Study, risk of an aneurysm increased threefold with any smoking history.127,148 Risk of AAA development further increases with number of cigarettes smoked, duration of smoking, and lack of filtration, indicating a dose-response relationship.149 In the Whitehall study of 18,403 male civil servants examined at age 40 to 64 years, aneurysm frequency increased from sixfold with manufactured cigarettes with filters to 25-fold with hand-rolled cigarettes.150 Smoking cessation can reduce the risk of aneurysm formation, with former smokers having a lower AAA risk than current smokers.136,149,151
Risk factors for cardiovascular disease in general (e.g., hypertension, hyperlipidemia) also increase the risk of AAA formation but are less potent risk factors than age, gender, and smoking.127,136,152–154 In the REACH registry, there was a clear modest association of both hypertension and hyperlipidemia with AAA.155 Earlier studies had suggested a less consistent relationship with hypertension,138,156 but aneurysm formation seems to correlate best with diastolic blood pressure157 or use of an antihypertensive medication.140 Some data suggest that hypertension may be a greater risk factor for aneurysm rupture than for initial aneurysm formation.158 The association between cholesterol and AAA is clear, although hyperlipidemia is a less potent risk factor than those mentioned previously.135,137,159 Risk of AAA formation increased 30% per 40 mg/dL total cholesterol in the Chicago Heart Association Detection Project in Industry cohort.138 For specific components of the lipid profile, higher levels of low-density lipoprotein (LDL) and lower levels of high-density lipoprotein (HDL) cholesterol are both associated with aneurysm formation.133,159 Similarly, the presence of atherosclerosis increases risk of aneurysm formation,133,134,137 although as mentioned previously, aneurysm formation is likely a process distinct from atherosclerosis. In the ADAM study of more than 100,000 subjects, hypertension, elevated cholesterol, and presence of other vascular disease increased risk of aneurysm formation by 15%, 44%, and 66%, respectively.127 In contrast, diabetes and black race appear protective against formation of an aneurysm.127,132,136,160 Diabetes decreases risk of aneurysm formation by 30% to 50%.136,157
A dramatic increase in frequency of AAA formation in relatives of patients with aortic aneurysm suggests a genetic component to the disease. Although cigarette smoking numerically accounts for the vast majority of AAAs in the population,136 the most potent risk factor for aneurysm formation is a history of aneurysm in a first-degree relative. Norgaard et al.161 identified an 18% incidence of aneurysms in first-degree relatives of patients with AAA. In the ADAM study, a family history doubled the risk of AAA, but was reported in only 5.1% of more than 100,000 participants.162 Investigations specific to the impact of family history demonstrate a larger risk. Several studies have demonstrated that a family history of AAA increases the risk of AAA four- to fivefold.157,163 Family history of AAA was also related to earlier AAA formation and rupture by nearly a decade.164 Rate of rupture was nearly fourfold higher in patients with a family history than in sporadic AAA patients. Frydman et al.165 screened the siblings of 400 AAA patients and found an AAA in 43% of male siblings and 16% of female siblings. More specifically, the risk of AAA formation consistently rises above 20% for men older than 50 who have a first-degree relative with AAA.161,166–169 Using segregation analysis, Majumder et al.170 reported that the relative risk of developing an AAA is 3.97 and 4.03 with paternal and maternal history, respectively. Risk increases to nearly 10-fold with an affected male sibling and 23-fold when a female sibling is affected.170 Twin studies further support a genetic component to AAA formation, with one study reporting an odds ratio (OR) of 71 (95% confidence interval [CI], 27-183) for monozygotic twins and 7.6 (95% CI, 3.0-19) for dizygotic twins.171
Despite the wealth of data supporting a genetic component to AAA formation, no clear mode of inheritance and no single candidate gene has been identified. Early studies suggested evidence of both sex-linked and autosomal dominant patterns of inheritance. Associations have been made with blood types, haptoglobin variations, α1-antitrypsin, and human leukocyte antigen class II (HLA-II) immune response genes.172–174 More than 100 reports on genetic associations with AAA have appeared in the literature,175 including genes related to cardiovascular disease, inflammation, and related signaling pathways. One study suggested an association between reduced AAA growth and five single-nucleotide polymorphisms (SNPs) in latent TGF-β binding protein (LTBP4), as well as an allelic variant of TGFB3.176 However, another study showed no association between genetic polymorphisms in the main receptors for TGF-β and AAA formation.177 Two genome-wide association studies (GWAS) have suggested an association between AAA and a SNP located on chromosome 3p12.3 in a region near the gene CNTN3 for contactin-3, a lipid-anchored cell adhesion molecule.178 Another GWAS in a population from Iceland and the Netherlands found a SNP on 9q33 associated with AAA with an OR of 1.21. The same SNP has been associated with coronary artery disease (CAD), peripheral artery disease (PAD), and pulmonary embolism (PE).179 The SNP resides in the gene coding for DAB2IP, a gene encoding a cell growth and survival inhibitor. In addition, the same gene variant associated with myocardial infarction (MI) at locus 9p21 has been associated with AAA.180,181 Finally, of interest but of unclear significance is the association between telomere length and AAA in a small cohort in the United Kingdom.182 This may represent the association of aging, telomere length, and aneurysm formation, but the direct pathophysiological link remains unclear.
Prognosis
The natural history of AAA is one of silent coexistence and sudden lethal rupture. In a large autopsy study performed over a quarter of a century, one fourth of abdominal aneurysms were ruptured on postmortem examination.126 The frequency of rupture was dependent largely on size in this study population, ranging from 9.5% in aneurysms smaller than 4 cm to 45.6% in aneurysms 7.1 to 10 cm in diameter. In a comparison of patients with aortic aneurysms divided into two groups at a cutoff point of 6 cm, survival was markedly decreased in the patients with larger aneurysms.183 In a single-center study that included 60 AAA ruptures over a 30-year period, only 2 occurred in patients with an aneurysm diameter smaller than 5 cm.184 Similarly, in a study in Rochester, Minnesota, no ruptures occurred in aneurysms smaller than 5 cm, whereas rupture occurred in 25% of AAAs larger than 5 cm.185 In 198 patients with aneurysms 5.5 cm in diameter or larger, but who were deemed too risky for surgery, 23% had presumed rupture over a mean follow-up of 1.6 years.186 Mortality rates as high as 64% are observed in patients who present with a ruptured aortic aneurysm.187
On the basis of these data, two large trials have been conducted to determine whether early recognition and treatment can alter the natural history of AAA. The U.K. Small Aneurysm Trial randomized 1090 patients aged 60 to 76 years with asymptomatic AAAs 4 to 5.5 cm in diameter to undergo early elective open surgery or ultrasonographic surveillance.188 In the surveillance group, surgery was performed when the aneurysm reached 5.5 cm. Early surgery did not affect overall mortality. At 3 years, nearly 20% of both groups had died, although abdominal aneurysms accounted for only a quarter of the deaths in both groups. Cardiovascular mortality unrelated to the aneurysm accounted for 40% of total mortality, and cancer caused slightly more than 20% of the deaths. In a similar study performed by the U.S. Veterans Administration, 1136 subjects aged 50 to 79 years with asymptomatic AAA 4 to 5.5 cm in diameter were randomized to undergo either early elective open surgery or ultrasonographic surveillance153 (Fig. 37-5). After 5 years, there was no significant difference in survival between the groups, each with a near 25% mortality rate. In this study, aneurysm-related deaths accounted for only 3% of total mortality. More recently, the PIVOTAL study demonstrated no difference between surveillance and early endovascular repair in patients with small aneurysms (measuring 4-5 cm), and no difference in aneurysm-related death in the two groups after 3 years of follow-up.189 Together, these data suggest that early repair of small aneurysms does not alter outcomes.
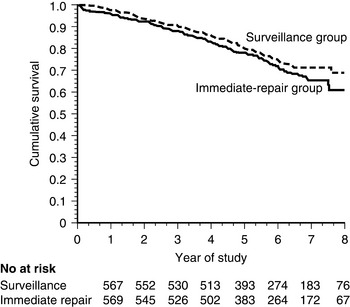
No significant difference in mortality was found between the two groups.
(From Lederle FA, Wilson SE, Johnson GR, et al: Immediate repair compared with surveillance of small abdominal aortic aneurysms. N Engl J Med 346:1437, 2002.)153
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


