Systemic arterial hypertension is one of the most common cardiovascular diseases of industrialized populations. It affects approximately 20% of adults in these societies, and a much higher proportion in certain demographic groups (e.g. blacks, the elderly). The disease is the major treatable risk factor underlying coronary heart disease (Table 1.1), and exacerbates and accelerates the atherosclerotic process. Moreover, hypertension is a key determinant risk for premature cardiovascular morbidity and mortality endpoints (Table 1.2). It is, therefore, necessary to understand the nature of the disease pathophysiologically; by doing so, it is then possible to conceive, develop, and select. This, then, is the mission of this atlas.
Unresolved (as to whether treatment reverses risk)
Left ventricular hypertrophy
Hyperinsulinism
Hyperuricaemia
Indices of inflammation (e.g. C-reactive protein)
Table 1.2 Complications and end-points promoted by hypertension that result in premature cardiovascular morbidity and mortality
Brain
Haemorrhagic stroke
Thrombotic stroke
Embolic stroke
Heart
Angina pectoris involving coronary arterioles
Occlusive epicardial coronary arterial diseases
Congestive heart failure
Left ventricular diastolic dysfunction
Kidney
Renal arterial disease
End-stage renal disease
Embolic renal disease
Exacerbation of diabetic renal disease
Other hypertensive emergencies
Dissecting aortic aneurysm
Accelerated and malignant hypertension
Crisis from phaeochromocytoma
Eclampsia
Other pressor emergencies
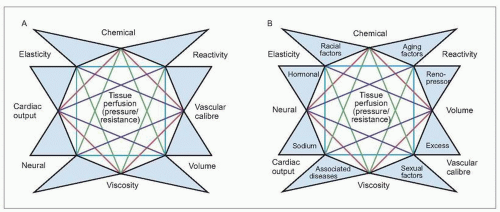
The mosaic
It was approximately 60 years ago that Irvine H. Page described his concept of the mosaic of hypertension (see Further reading). Inherent in his thesis was the belief that hypertension is multifactorial in causation. This is because all of the mechanisms that serve to control arterial pressure in normal individuals as well as in those patients with hypertensive disease relate to each other in a kaleidoscopic fashion, each with the others. Thus, all mechanisms are critical for maintaining homeostasis, physiologically or pathophysiologically (1.1A, B).
The factors depicted in Page’s mosaic clearly are not allinclusive but serve to satisfy the underlying model suggesting that many (if not most) diseases are multifactorial in causation. In the case of hypertension, the fundamental driving physiological purpose is to maintain normal tissue perfusion. In hypertension, this is accomplished at the expense of an increased vascular resistance and, hence, the elevated arterial pressure which is the primary clinical characteristic of hypertensive disease.
1.1 Page’s mosaic theory of hypertension. A: the original theory; B: including additional factors as modified by the author. Each of these (and other) factors interrelate with one another in order to maintain normal tissue perfusion in response to an increasing vascular resistance and at the expense of the abnormally elevated arterial pressure. (Modified from Frohlich ED: Clinical classifications of hypertensive diseases. In: Atherosclerosis and Coronary Artery Disease. V Fuster, R Ross, E Topol (eds.). Lippincott-Raven, Philadelphia, 1996.)
Altered haemodynamics
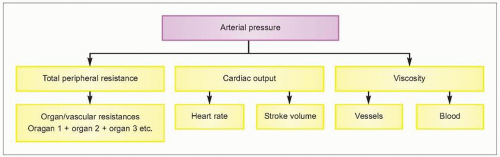
To understand the pathophysiological alterations associated with the systemic arterial hypertensive diseases, there must first be a clear-cut understanding of the haemodynamic alterations associated with a persistent elevation of arterial pressure. By definition, hypertension is a haemodynamic disorder in which the elevated arterial pressure may be associated with an increased cardiac output and/or total peripheral resistance (1.2).
In most patients with essential hypertension, the elevated arterial pressure is associated with an increased total peripheral resistance. In some patients, however, an elevated cardiac output may also participate. The relationship between arterial pressure, cardiac output, and total peripheral resistance is discussed more extensively in subsequent chapters. However, when one considers the magnitude of the elevated pressure, changes in blood viscosity do not have major importance. Nevertheless, intravascular rheological changes may alter local tissue blood flow dynamics in the major target organs. Thus, it is possible that some degree of increased viscosity promotes changes in blood rheological characteristics which could exacerbate the haemodynamic alterations in the coronary, renal, and brain circulations.
For the most part, however, the increased total peripheral resistance (or, in organ circulations, their corresponding vascular resistances) is the established haemodynamic hallmark of the hypertension and is more or less uniformly distributed throughout the various organ circulations. The mechanism responsible for this resistance increase is an augmented vascular smooth muscle tone, primarily in the precapillary arterioles, and accounts for the state of increased arteriolar tone (i.e. arteriolar constriction) that is implicit in the multifactorial nature of the disease (Table 1.3).
Table 1.3 Active and passive mechanisms that alter vascular resistance
I. Constriction
Active
Adrenergic stimulation (i.e. increased neural input or increased vascular responsiveness to normal neural input)
1.2 The haemodynamic concept inherent in hypertension.
This book emphasizes those mechanisms that have been related to essential hypertension, since this primary form of hypertension occurs in approximately 95% of all patients with systemic arterial hypertension. Moreover, this pathophysiological discussion is also relevant to other (i.e. secondary) clinical forms of hypertension. Consideration of the pertinent underlying pressor mechanisms in those secondary forms of hypertension provides a basis for a more comprehensive insight into the overall mechanisms that could participate in patients with essential hypertension (Table 1.4).
Table 1.4 Classification of the various forms of systemic arterial hypertension
Primary (essential) hypertension (hypertension of undetermined cause)
Borderline (labile) or ‘high normal’ hypertension or prehypertension (essential hypertension)
Antidepressant therapy (tricyclic antidepressants, MAO inhibitors)
Chronic steroid administration
Cyclosporine (transplantation and certain disease immunosuppressive therapy)
Beta-adrenergic receptor agonists (e.g. for asthma)
Radiation nephritis and arthritis
Lithotripsy therapy for renal calculi
MAO, monoamine oxidase.
Arteriolar constriction
Arteriolar (and, for that matter, venular) smooth muscle tone is increased in hypertension, although all of the mechanism(s) responsible are not entirely known. No doubt, this relates to the many pressor and depressor factors that normally participate in regulating vessel tone and calibre and, hence, arterial pressure (Table 1.3). It follows that these factors also participate in the increased vascular resistance in most patients with essential hypertension. Lessons concerning regulation of increased vascular resistance have been learned from the variety of secondary forms of hypertension in which specific pressor and depressor mechanisms are involved (Table 1.4).
It is likely that the increased vascular resistance in most patients with essential hypertension may be mediated through more than one pressor mechanism. Some of these mechanisms are predetermined by inborn genetic factors, since it has become increasingly apparent that the pathophysiological alterations in essential hypertension are polygenetic in origin. Furthermore, many of the pressor and depressor mechanisms that seem to be operative have been well documented to increase actively vascular smooth muscle tone. Many new mechanisms are elucidated with each passing year. Thus, vascular smooth muscle tone is abnormally increased as a result of one or more of those factors that participate in the underlying disease process and are then expressed in the clinical manifestations of that patient’s disease. As a consequence of the increased total peripheral resistance, arterial pressure rises in order to maintain tissue perfusion; this occurs at the expense of the vascular and cardiac systems, and the specific indices of organ damage and functional impairment that secondarily result.
As suggested in Tables 1.2 and 1.3, the increased tone of the arteriolar or venular smooth muscle occurs no matter what mechanism(s) participate. Thus, for example, the vascular myocyte is constricted by enhanced adrenergic input or elevated circulating levels of catecholamines; alterations in circulating or local autocrine/paracrine effects of humoral substances; local or systemic participation of vasoactive peptides (e.g. angiotensin II, endothelin); ions; and growth factors. Alternatively, increased vascular resistance may also be produced by reduced local or systemic amounts of vasodilating agents, local vasoactive peptides or ions, and vasoactive metabolites (Table 1.3). Whatever the myocytic stimulus, there is a resultant rise in cytoplasmic free calcium ions from their resting state that results in enhanced phosphorylation of myosin light chains. This increased calcium ionic milieu may be achieved either through an inflow of calcium ions through calcium- or other receptor-activated membrane channels or by a release of calcium ions from intracellular organelles, although calcium may be released from the mitochondria or from binding with protein substrates through secondarily activated biochemical processes. The net increase in intracytoplasmic calcium ion concentration promotes the formation of inositol triphosphate (IP3) and diacylglycerol. IP3 serves as the second messenger, mediating the calcium ion release and the resulting mechanical coupling that permits an enhanced state of contractility of vascular smooth muscle.
Arteriolar structure
Another factor participating in the increased vascular resistance of hypertension is an increased wall-to-lumen ratio of the arteries and arterioles. This structural alteration in hypertension serves to amplify the arteriolar responsiveness to constrictor stimuli that maintains the hypertensive disease process. Recent investigations have suggested that the haemodynamic stress of vessel stretch may be an important additional mechanism responsible for the vessel wall thickening, or even of myocytic hypertrophy of the left ventricle. Several reports have indicated that upon stretch of the ventricular or arteriolar (e.g. renal, coronary) myocyte, one or more of a vast array of ‘early genes’ or protooncogenes participate in initiating DNA-directed myocytic and collagen (and likely other) growth. Some of these growth factors are themselves vasoconstrictors (e.g. angiotensin II, norepinephrine [noradrenaline], endothelin), and they may even be generated within the arteriolar or ventricular endothelium or wall itself. Intriguingly, they may also participate in the separate but related process of atherogenesis. Hence, this may explain the close relationship of these two common and comorbid diseases (i.e. hypertensive vascular disease and atherosclerosis).
Only gold members can continue reading. Log In or Register to continue