Chapter 34 Pathophysiology, Clinical Evaluation, and Medical Management of Aortic Dissection
Acute aortic dissection is an uncommon but life-threatening emergency that requires prompt diagnosis, rapid triage, and immediate medical, endovascular, or surgical treatment. A unified effort across several international centers over the past 15 years has led to the establishment of a detailed registry that describes major aspects of presentation, management, and outcomes of patients with acute aortic dissection.1 This longitudinal experience has given new clinical insights into an old disease and spawned additional multicenter efforts to explore its genetics and pathobiology. Although gains have been made in the delivery of life-saving care to patients with acute aortic dissection, hospital mortality rates remain distressingly high. Enhanced awareness of risk factors for aortic dissection, presentation features, diagnostic pathways, and medical, endovascular, and surgical treatment strategies is a critical first step toward improving outcomes.
Epidemiology
Published figures on the incidence of aortic dissection likely underestimate the actual occurrence rate, since misdiagnosis of this condition is common and the percentage of acute aortic dissection patients who expire before hospital presentation cannot be accurately estimated.2 Nevertheless, acute aortic dissection is a rare event. Analysis of the Swedish National Cause of Death Register between 1987 and 2002 estimated the incidence of thoracic aortic aneurysm or dissection to be 16.3 per 100,000 men and 9.1 per 100,000 women; others have reported acute aortic dissection to affect 30 in 1 million individuals per year.3 By comparison, acute myocardial infarction (AMI) is 140-fold more common.1 Data from the International Registry of Aortic Dissection (IRAD) indicate that the mean age of patients at presentation with aortic dissection is 63 years, with men accounting for 63% of cases.1
Classification
Classifying aortic dissection according to anatomical location and time from onset of symptoms helps stratify risk and guide selection of initial treatment strategy (Fig. 34-1). The Stanford classification system designates dissections that involve the aorta proximal to the brachiocephalic artery (i.e., root and ascending aorta) as type A, and those that do not as type B.4 This distinction is clinically important because dissection involving the ascending aorta is a key determinate of early death and major morbidity. Location of the intimal tear does not influence Stanford dissection type. In the older DeBakey classification scheme, a type I dissection originates within the ascending aorta and extends for a variable distance beyond the take-off the innominate artery. A DeBakey type II dissection is confined to the ascending aorta, and a type III dissection originates in the descending thoracic aorta beyond the origin of the left subclavian artery and terminates above (type IIIA) or extends below (type IIIB) the level of the diaphragm.5 Although there is no single universally accepted classification system, the Stanford classification scheme is most often used in practice today and will be used throughout this chapter. The terms communicating and noncommunicating refer to the presence or absence, respectively, of blood flow between the true and false lumens of the aorta.
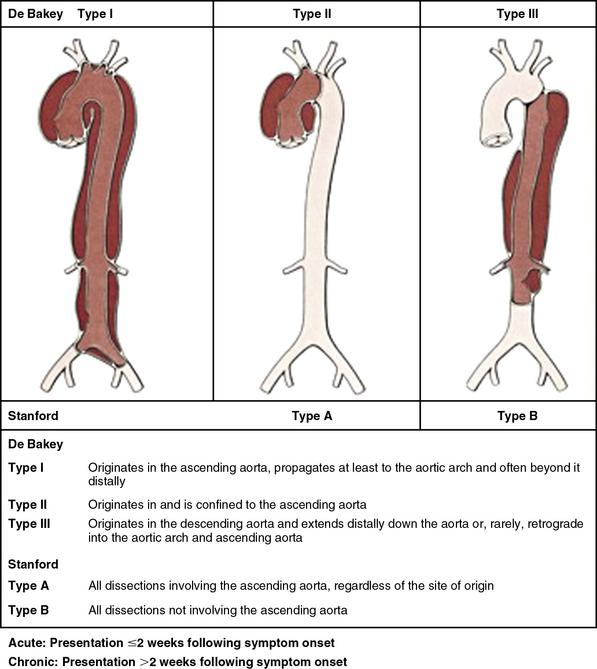
Figure 34-1 Aortic dissection type according to De Bakey and Stanford classification systems.
(From Nienaber C, Eagle KA: Aortic dissection: new frontiers in diagnosis and management. Part I: from etiology to diagnostic strategies. Circulation 108:628–635, 2003.)7
Pathogenesis
Forces that weaken the medial layer of the aorta increase the probability of dilation, aneurysm formation, and dissection (Box 34-1). Acquired and genetic diseases that mediate this process are discussed later. In classic acute aortic dissection, the initiating event is an intimal tear through which blood rapidly surges distally into the media under systolic pressure, splitting the layers of the aortic wall and creating an intimal flap that separates the true from the false lumen.
 Box 34-1
Box 34-1Intimal Tear
Contemporary imaging modalities or autopsy findings identify the primary entry tear in approximately 90% of cases. It is most frequently located a few centimeters above the level of the aortic valve along the greater curvature of the aorta in cases of type A dissection and accounts for nearly 60% of all cases. Compared with other locations in the ascending aorta, the proximal few centimeters of the greater curvature are exposed to relatively greater hemodynamic, shear, and torsional force. A pivot region located in the descending thoracic aorta just beyond the insertion of the ligamentum arteriosum where the relatively mobile arch meets the fixed descending thoracic aorta is the second most common entry site for intimal tears, which will then propagate as a type B dissection (30% of cases). Arch entry occurs in 7% of cases. The abdominal aorta is the least common site for entry (3% of cases), despite the high prevalence of intima media ulcers in patients with atherosclerotic disease in this segment.6–8
The dissecting hematoma usually propagates in an anterograde direction, although retrograde extension can occur. By this mechanism, as many as 20% of dissections that originate in the distal arch or descending thoracic aorta may involve the ascending aorta.9 In rare cases, a second tear may occur, resulting in a three-channel dissection10 (Fig. 34-2).
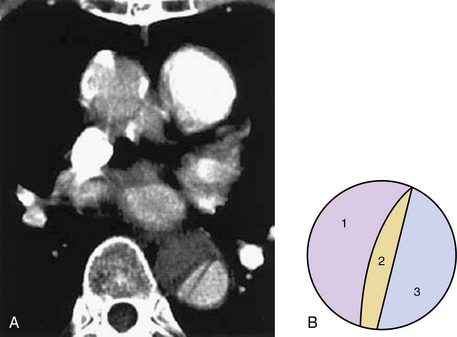
Figure 34-2 Three-channeled aortic dissection.
(From Yoshida S, Shidoh M: Three channeled aortic dissection. Postgrad Med J 79:532, 2003.)
Aortic Rupture and End-Organ Malperfusion
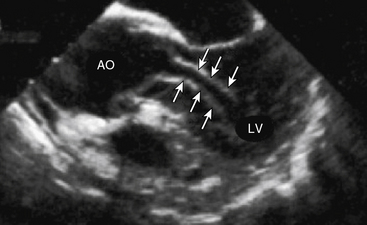
Aortic rupture, defined as tearing in the vessel wall that results in extravascular hemorrhage, most commonly occurs with trauma (e.g., motor vehicle collision) but may occur as a complication of the primary dissection.11 Rupture into the pericardial space resulting in cardiac tamponade occurs in type A dissection, whereas rupture into the left pleural space is usually encountered with type B dissection. Dissection-mediated end-organ ischemia or infarction occurs from (1) mechanical compression of aortic branch vessels by false lumen hematoma, (2) extension of the dissection plane across the ostium of the branch vessel, or (3) dynamic vessel inlet obstruction caused by an oscillating intimal flap. Compromise of coronary, brachiocephalic, mesenteric, renal, spinal, and iliac circulations can occur and result in a myriad of clinical presentations. Occlusion of the left ventricular (LV) outflow tract by an intimal flap has been reported (Fig. 34-3).
False Lumen Thrombosis
Thrombosis of blood within the false lumen may seal the entry tear, thus eliminating communication with the true lumen and interruption of false lumen expansion. Partial thrombosis of the false lumen, however, has been identified as a risk factor for long-term death in patients with type B dissection.12,13 Elevation of pressure with partial thrombosis within the false lumen may lead to further extrinsic compression of the true lumen and impairment of blood flow to critical organs. Alternatively, it has been proposed that partial thrombosis of the false lumen is associated with worse clinical outcomes by promoting vascular inflammation, hypoxia, and/or neovascularization with weakening of adjacent vascular structures and an increased risk for aortic rupture.13–17
Persistent patency of the false lumen is also associated with a higher risk of long-term complications such as late rupture or false aneurysm formation requiring operative intervention.12,15–17 Although native aortic aneurysm disease is often a risk factor for dissection, a dissection need not result in aneurysm formation. The term “dissecting aneurysm” is an inaccurate anachronism, and these diseases are not synonymous, a distinction that is particularly important when considering their natural histories and treatments.
Predisposing Genetic Factors
As is true for aneurysms, any process that leads to destruction or degeneration of the major supporting elements of the aortic media (elastin, collagen, smooth muscle cells [SMCs]) can predispose to development of dissection. The histopathological term medial degeneration refers to noninflammatory destruction or fragmentation of elastic lamellar units, dropout of SMCs, and accumulation of mucopolysaccharide ground substances (not always in distinct cystic spaces), which characterize the final common pathway for a variety of processes that affect the integrity of the aortic media (Fig. 34-4).
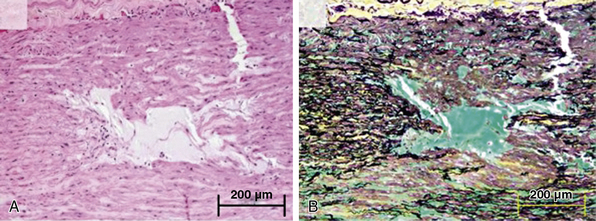
Figure 34-4 Cystic medial degeneration.
(From Maleszewski JJ, Miller DV, Lu J, et al: Histopathologic findings in ascending aortas from individuals with Loeys-Dietz syndrome [LDS]. Am J Surg Pathol 33:194–201, 2009.)
Marfan’s syndrome (MFS) is the most common inherited connective tissue disorder, with an estimated prevalence of 1 case per 3000 to 5000 individuals irrespective of racial, ethnic, or geographic considerations.18–21 If untreated, aortic disease in MFS is progressive and incompatible with normal longevity. Half of patients in whom aortic dissection occurs at younger than 40 years of age have a history of MFS.22 Mutations in the gene encoding fibrillin-1 (FBN1), a critical component of the microfibrils that help form cellular adhesions in the extracellular matrix (ECM), are largely responsible for disease expression.20 Medial degeneration is not pathognomic of MFS and may be present in numerous other conditions.23
Marfan’s syndrome disease expression is heterogenous, but the presence of an aortic root aneurysm (aortic diameter z score ≥2) and ectopia lentis are sufficient to make the diagnosis even in the absence of a family history24 (Box 34-2A). Conversely, in the absence of either of these two clinical features, the revised Ghent nosology for MFS outlines a scoring system based on the presence of FBN1 mutation and/or other key systemic features of MFS to make the diagnosis (Box 34-2B). Systemic features suggestive of MFS include positive wrist sign (entire distal phalanx of adducted thumb extends beyond ulnar border of the palm), positive thumb sign (tip of thumb covers entire fingernail of fifth finger when wrapped around contralateral wrist), pectus excavatum, pneumothorax, dural ectasia, hindfoot deformity, and protrusio acetabuli. Other clinical features less strongly associated with MFS include mitral valve prolapse, various abnormal facial features, and thoracolumbar kyphosis.
 Box 34-2A Revised Ghent Criteria for Diagnosis of Marfan’s Syndrome and Related Conditions*
Box 34-2A Revised Ghent Criteria for Diagnosis of Marfan’s Syndrome and Related Conditions*
In the Absence of Family History
In the Presence of Family History
(5) EL and FH of MFS (as defined above) = MFS
(6) Syst (≥7 pts) and FH of MFS (as defined above) = MFS†
(7) Ao (z ≥2 above 20 years old, ≥3 below 20 years old) + FH of MFS (as defined above) = MFS†
Ao, aortic diameter at the sinuses of Valsalva above indicated z-score or aortic root dissection; EL, ectopia lentis; ELS, ectopia lentis syndrome; FBN1, fibrillin-1 mutation; FBN1 not known with Ao, FBN1 mutation that has not previously been associated with aortic root aneurysm/dissection; FBN1 with known Ao, FBN1 mutation that has been identified in an individual with aortic aneurysm; MASS, myopia, mitral valve prolapse, borderline (z <2) aortic root dilation, striae, skeletal findings phenotype; MFS, Marfan’s syndrome; MVPS, mitral valve prolapse syndrome; Syst, systemic score (see Box 34-2B); z, z-score.
* In general, MFS is diagnosed in the presence of aortic root dilation/dissection and ectopia lentis; aortic root dilation/dissection plus FBN1 mutation; aortic root dilation plus sufficient systemic findings (Box 34-2B, ≥7 points); ectopia lentis plus FBN1 mutation previously associated with aortic disease; or in an individual with a positive family history of MFS, the diagnosis is made in the presence of ectopia lentis, or a systemic score ≥7 points, or aortic root dilation.
† Caveat: without discriminating features of Shprintzen-Goldberg’s syndrome, Loeys-Dietz’s syndrome, or vascular Ehlers-Danlos’ syndrome and after TGFBA1/2, collagen biochemistry, COL3A1 testing if indicated. Other conditions/genes will emerge with time.
 Box 34-2B Scoring of Systemic Features
Box 34-2B Scoring of Systemic Features
Wrist and thumb sign: 3 (wrist or thumb sign: 1)
Pectus carinatum deformity: 2 (pectus excavatum or chest asymmetry: 1)
Hindfoot deformity: 2 (plain pes planus: 1)
Reduced US/LS and increased arm/height and no severe scoliosis: 1
Scoliosis or thoracolumbar kyphosis: 1
Facial features (3/5): 1 (dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia)
mAXIMUM tOTAL: 20 points; score ≥7 indicates systemic involvement.
US/LS, upper segment/lower segment ratio.
From Loeys BL, Dietz HC, Braverman AC, et al: The revised Ghent nosology for the Marfan’s syndrome. J Med Genet 47:476–485, 2010.24
Using an in vivo murine model of MFS, Dietz et al. demonstrated that fibrillin-1 is a key target for binding of transforming growth factor (TGF)-β, a molecule involved in activation of inflammatory, fibrotic, and metalloproteinase cell signaling pathways.25 To investigate the possibility that angiotensin I (AT1)-mediated TGF-β activation could represent a pharmacological target to modify development of aortic dilation in MFS, Habashi et al. studied the effects of losartan, an AT1 receptor antagonist, on aortic aneurysm formation in transgenic mice encoding a cysteine-to-glutamine substitution at position 1039 in the fibrillin-1 gene (Fbn1C1039G/−). Mice with this fibrillin-1 mutation, the most common class of mutation associated with MFS, demonstrate significant and progressive aortic root dilation compared with wild-type mice.26 Treatment with losartan attenuated TGF-β signaling in the aortic wall and resulted in full normalization of aortic wall thickness and a marked improvement in aortic wall architecture (i.e., a decrease in elastin fiber disruption) compared with placebo or β-adrenergic receptor antagonist therapy with propranolol.
These landmark observations have advanced understanding of the molecular basis of MFS and provided new targets for therapy. In one small clinical trial in young MFS patients, initiation of losartan resulted in a significant decrease in rate of growth of the aortic root (mean change 0.46 ± 0.62 mm/yr vs. 3.5 ± 2.8 mm/yr prior to treatment; P < 0.001).27 These findings were supplemented by Ahimastos et al., who conducted a randomized placebo-controlled clinical trial testing the effect of the angiotensin-converting enzyme inhibitor (ACEI) perindopril on aortic root diameter and aortic stiffness in 17 MFS patients.29 At 24 weeks, circulating TGF-β levels and central pulse wave velocities were lower and aortic root diameters were smaller in perindopril-treated patients. Several larger randomized clinical trials directed at modulating TGF-β signaling are ongoing (clinicaltrials.gov; NCT00683124, NCT00782327 NCT00429364, NCT00723801).
Limited forms (i.e., forme frustes) of MFS that feature cardiovascular manifestations include mitral valve prolapse syndrome (MVPS) (mitral valve prolapse, pectus excavatum, scoliosis, and mild arachnodactyly) and the MASS phenotype (myopia, mitral valve prolapse, borderline and nonprogressive aortic root dilation, skeletal findings and striae). There is increasing awareness of familial thoracic aortic aneurysm disease (FTAAD), though candidate genes affecting both matrix and SMC components have been identified in only 20% of such patients. Vascular-type Ehlers-Danlos’ syndrome (EDS) is associated with arterial rupture and dissection, including the aorta.24,28
Ehlers-Danlos’ syndrome (1:5000 births) comprises a heterogeneous group of disorders characterized clinically by hypermobile joints, hyperextensible skin, tissue fragility, and a predisposition to spontaneous vascular rupture. Aortic involvement occurs in EDS type IV, an autosomal dominant disorder attributed to structural defects in the pro-α1 (III) chain of type III collagen, encoded by the COL3A1 gene on chromosome 2q31.30
FTAAD has been mapped to other genetic loci, including 16p13.11 (MYH11 gene), 5q13-14, and 11q23.2-q24, which are not associated with abnormalities of fibrillin or collagen.31,32 More than five mutations in the fibrillin-1 gene have been identified in patients with familial or spontaneous thoracic aortic aneurysm and dissection, with histopathological changes characteristic of medial degeneration, yet with no demonstrable abnormalities of collagen or fibrillin in fibroblast culture.33,34 Polymorphisms encoding the gene vitamin K epoxide reductase complex subunit 1 (VKORC1), which results in under-carboxylation of specific matrix proteins, are associated with calcification of the arterial wall. In patients carrying the C allele (CT or CC), a relative two-fold increase in the probability of developing aortic dissection has been observed.35 A missense mutation in the ACTA2 gene that encodes for actin filaments in SMCs is linked to 14% of patients with FTAAD.36 As a consequence of this mutation, intracellular actin filament assembly is disrupted, promoting focal areas of SMC disarray, decreased SMC contraction, and medial degeneration of the aorta. This phenotype is felt to be associated with aortic wall weakening and increased predisposition to dissection.
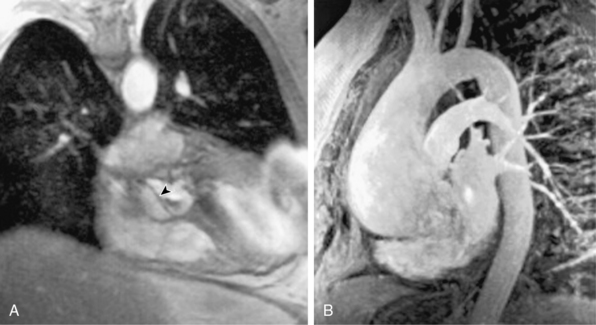
Bicuspid aortic valve (BAV) disease is the most common congenital cardiac anomaly in adults (4:1000 live births) and is often accompanied by an aortopathy that is histologically similar but less severe than that observed in patients with MFS. Similar pathological changes have been described in patients with aortic coarctation (many of whom have BAV disease), Noonan’s syndrome, Turner’s syndrome, and polycystic kidney disease. Dilation of the root and (more commonly) the ascending aorta is present in up to 40% of patients with BAV disease and is a risk factor for dissection or rupture (Fig. 34-5).
Acquired Disorders
Systemic hypertension is the most common treatable risk factor for aortic dissection and is present in approximately 75% of patients.2 Hypertension accelerates the normal aging process and leads to intimal thickening, SMC apoptosis, fibrosis, loss of elasticity, and compromise of nutritive blood supply. Decreased aortic compliance and vulnerability to pulsatile forces predispose to injury and create a substrate for dissection.
Iatrogenic Dissection
In the IRAD registry, iatrogenic aortic dissection after cardiac surgery or catheterization accounted for 5% of the total reported.37 Older age, hypertension, and severe peripheral vascular disease are risk factors associated with procedure-related dissection.38 Pain may be absent in iatrogenic dissection. Retrograde dissections created at the time of catheterization usually seal spontaneously on withdrawal of the catheter. Aortic atherosclerotic plaques may prevent longitudinal propagation of a dissection. Dissections arising from sites where the aorta has been incised or cross-clamped may occur intraoperatively or at any time following surgery. Deceleration injury from high-speed accidents results in aortic transection with false aneurysm formation and rupture, most commonly in the region of the aortic isthmus just beyond the origin of the left subclavian artery. Transection results in a transmural tear that is different both pathologically and etiologically from aortic dissection.
Dissection in Pregnancy
Aortic dissection as a complication of pregnancy is rare, although by some estimates 50% of all dissections in women younger than 40 occur during labor, delivery, or early after childbirth.39 Histopathological changes affecting the aortic media of pregnant women have been described, including alterations in elastic fibers and SMCs.40 Both estrogen and relaxin are associated with alterations in matrix metalloproteinase (MMP) homeostasis and contribute to vascular remodeling and a susceptibility to injury independent of the hemodynamic stress of labor and delivery. In many cases, pregnancy unmasks primary conditions that predispose to aortic dissection (e.g., MFS). In those patients with preexisting MFS or BAV disease, aortic root size greater than 4.0 cm is a contraindication to pregnancy, owing to increased risk for spontaneous rupture or dissection.2
Drug Use and Other Acquired Conditions
Recent cocaine use, particularly among young men who smoke tobacco, is an additional risk factor for aortic dissection.41 Among 38 patients with acute aortic dissection occurring over a 20-year period in an urban center, 37% reported cocaine (in particular, crack cocaine) use within the preceding 24 hours (mean 12 hours). Chronic amphetamine use and/or dependence appear to increase the probability of developing a thoracoabdominal aortic dissection in those aged 18 to 49 years.42 Presumed mechanisms for aortic injury from cocaine and amphetamine use involve oxidant stress–mediated endothelial dysfunction and extreme catecholamine-induced shear forces, with abrupt hypertension and tachycardia that collectively lead to weakening of the aortic media and predisposition to tearing.
Inflammatory diseases of the aorta can lead to destruction of ECM proteins and SMCs, with subsequent aneurysm formation and/or dissection. Aortic dissection has been reported in patients with Takayasu’s disease, giant cell aortitis, Behçet’s disease, relapsing polychondritis, systemic lupus erythematosus (SLE), and the aortitis associated with inflammatory bowel disease.2 Syphilitic aortitis, on the other hand, does not predispose to dissection, perhaps because of intense reactive medial scarring and fibrosis that occurs in response to the spirochetal infection. Pheochromocytoma and weight lifting (believed due to intense or repetitious Valsalva maneuvers) also predispose to aortic dissection.
Clinical Presentation
The most important element of any diagnostic algorithm for suspected acute aortic syndrome is a high clinical index of suspicion, based foremost on the presenting history and physical examination (Box 34-3). Absent an appreciation for the cardinal features of dissection, the diagnosis can be missed in a substantial number of patients. Simple clinical prediction rules have been developed to estimate probability of acute aortic dissection.43 The IRAD investigators have recently confirmed the sensitivity of 12 clinical risk markers proposed in the 2010 American College of Cardiology Foundation/American Heart Association (ACCF/AHA) thoracic aortic disease guidelines2 (Table 34-1). These markers, assessed at bedside, were divided into three distinct categories: predisposing factors, characteristics of the pain at time of presentation, and key physical examination findings.44 The presence of risk factor(s) from at least one category identified 95.7% of acute aortic dissection patients in the IRAD database.
 Box 34-3 Acute Aortic Syndromes
Box 34-3 Acute Aortic Syndromes
Adapted from Hiratzka LF, Bakris GL, Beckman JA, et al: 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease. J Am Coll Cardiol 55:e27, 2010.2
Table 34-1 Percentage of Patients in the International Registry of Acute Aortic Dissection (1996–2009) with Each of 12 High-Risk Clinical Markers Observed at Time of Presentation with Acute Aortic Dissection*
| Aortic Dissection Detection Risk Category | Clinical Characteristics | % of Patients (N = 2538) |
|---|---|---|
| 1 | MFS | 4.3 |
| 1 | Family history of aortic disease | 1.9 |
| 1 | Known aortic valve disease | 11.9 |
| 1 | Recent aortic manipulation | 2.8 |
| 1 | Known thoracic aortic aneurysm | 14.7 |
| 2 | Abrupt onset of pain Severe pain intensity | 79.3 72.7 |
| 2 | Ripping or tearing pain | 21.7 |
| 3 | Pulse deficit or systolic blood pressure differential | 20.3 |
| 3 | Focal neurological deficit (in conjunction with pain) | 10.8 |
| 3 | Murmur of aortic insufficiency (new or in conjunction with pain) | 23.6 |
| 3 | Hypotension/shock | 16.0 |
MFS, Marfan’s syndrome.
* The aortic dissection detection (ADD) score aims to enhance early diagnosis of acute aortic dissection. The ADD score is calculated by determining the number of categories in which any of 12 high-risk clinical features are present in patients with symptoms suggestive of acute aortic dissection. For example, in a patient with a family history of aortic disease (category 1) and known thoracic aneurysm (also category 1), the ADD score would be 1. Likewise, the ADD score is 2 in a patient with Marfan’s syndrome (category 1) and a blood pressure differential (category 3). A retrospective analysis of the International Registry of Acute Aortic Dissection determined that among 2538 patients with acute aortic dissection, 95.7% had an ADD score ≥1. The ADD score may therefore provide the clinician with a simple and effective bedside method to inform further diagnostic testing and/or treatment in patients with suspected aortic dissection. Importantly, the negative predictive value for acute aortic dissection in patients with an ADD score of 0 has not yet been established.
From Rogers AM, Hermann LK, Booher AM, et al: Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection on initial presentation. Circulation 123:2213–2218, 2011.44
History
Diagnosis of aortic dissection may be missed on initial clinical evaluation in about a third of cases, and an equal number are detected only at autopsy.1,44 Chest pain is the dominant feature of the clinical presentation and occurs in over 90% of patients.2 It is qualitatively severe and may in many cases be distinguished from coronary ischemia by abrupt onset and maximal intensity at inception. More than 84% of aortic dissection patients described chest pain as “worst ever” in the IRAD registry.2,45 The pain is characterized as sharp more often than tearing or ripping in nature, and may radiate or be sensed anteriorly (suggestive of type A dissection) or in the interscapular, lower back, or abdominal area (suggestive of type B dissection). Visceral discomfort or limb pain may be indicative of aortic branch vessel ischemia from malperfusion.
Syncope is a particularly ominous presenting symptom and may reflect cardiac tamponade from intrapericardial aortic rupture, cerebral malperfusion, and/or neurally mediated hypotension in response to the intense pain of the dissection. In the IRAD registry, patients with syncope were more likely to die in the hospital or suffer a stroke. Neurological complications are noted in up to 20% of aortic dissection patients. For example, paraplegia may develop when critical impairment of flow to the anterior spinal artery, thoracic intercostals, or the artery of Adamkiewicz occurs. Abdominal pain is an underrecognized symptom of acute aortic dissection; when present, it is associated with elevated in-hospital mortality and increased frequency of malperfusion syndromes.2,43,44
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


