LEARNING OBJECTIVES
Learning Objectives
The student will be able to define restrictive lung diseases and differentiate their various forms including etiology, pathogenesis if known, and clinical presentation.
The student will be able to describe and recognize gross and microscopic features of acute and chronic restrictive lung diseases.
Restrictive lung diseases are characterized by reduced lung compliance that requires greater pressure to inflate the lungs and, clinically, typically are manifest as dyspnea. Restrictive lung disease can result from external compression of the lung parenchyma; examples include severe scoliosis, chest wall tumors, and expansion of the pleural space by fluid or air (Chaps. 26 and 29). This chapter will focus on restrictive lung diseases in which the restriction is intrinsic to the lung rather than due to external compression. Although many different restrictive lung diseases will be discussed, there are some common themes to restrictive lung disease. Many such diseases show thickening of alveolar septa and alveolar epithelial and endothelial injury that lead to V̇A/Q̇ mismatch. With progression of many such diseases, patients develop severe hypoxemia and respiratory failure. These are often complicated with pulmonary hypertension and cor pulmonale (right ventricular dilatation due to lung disease).
ACUTE RESTRICTIVE LUNG DISEASES
The acute respiratory distress syndrome (ARDS) is a clinical syndrome characterized by the acute onset of respiratory distress with hypoxemia, reduced lung compliance, and diffuse pulmonary infiltrates in the absence of primary left heart failure; a less severe form of the syndrome is acute lung injury (ALI) (Chap. 28). Diffuse alveolar damage (DAD) is the morphologic counterpart of ALI/ARDS. Though DAD can complicate many conditions (Table 23.1), more than one-half of cases occur in the settings of sepsis, diffuse pulmonary infections, gastric aspiration, and trauma.
| Infection | Inhaled Irritants | Physical Injury |
|---|---|---|
| Sepsis | Oxygen toxicity | Mechanical trauma, head injury |
Diffuse pulmonary infections Viral pneumonia Mycoplasma pneumonia Pneumocystis pneumonia Miliary tuberculosis | Smoke Irritant gases Misc. chemicals | Pulmonary contusion Near-drowning Fractures with fat embolism Burns Ionizing radiation |
| Gastric aspiration | ||
| Chemical Injury | Hematologic Conditions | Hypersensitivity Reactions |
| Heroin or methadone overdose | Multiple transfusions | Organic solvents |
| Acetylsalicylic acid | Disseminated intravascular coagulation | Misc. drugs |
| Barbiturate overdose | ||
| Paraquat poisoning | ||
| Pancreatitis | Uremia | Cardiopulmonary Bypass |
The pathogenesis of DAD begins with endothelial damage or, less frequently, epithelial damage. Within 30 minutes, macrophages secrete canonical proinflammatory cytokines including TNF-α, IL-1, and IL-8, leading to neutrophil chemotaxis and activation (Chap. 10). Activated neutrophils secrete oxidants, proteases, platelet activating factor (PAF), and leukotrienes, the results of which are tissue damage, edema, inactivation of pulmonary surfactant, and the formation of hyaline membranes, the morphologic hallmark of DAD (see below). Later, macrophage secretion of transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF) causes proliferation of fibroblasts with subsequent synthesis of collagen. The pathogenesis of DAD is further discussed in Chap. 28.
Morphologically, lungs with DAD show reduced crepitus and resemble liver in consistency. Early in the course, the lungs are dense and dark red; as collagen deposition occurs, their color changes to gray (Fig. 23.1). Microscopically, DAD shows a spectrum of changes that can be organized into three phases (Fig. 23.2). The earliest of these is the exudative phase, with vascular congestion, interstitial and intra-alveolar edema, alveolar epithelial necrosis, neutrophil margination, dilatation and/or collapse of alveolar ducts, fibrin thrombi, and hyaline membranes. Hyaline membranes, the hallmark of DAD, are composed of edema fluid and necrotic epithelial cells. Subsequent to this exudative period is the proliferative phase, in which there is type 2 pneumocyte hyperplasia as well as fibroblast infiltration of the interstitium and the intra-alveolar exudate (ie, the hyaline membranes). Finally, as fibroblasts synthesize collagen, DAD enters the fibrotic phase, with fibrosis of the exudate (also described as organization) and expansion of the interstitium by fibrosis.
FIGURE 23.1
In diffuse alveolar damage (DAD), lungs are initially dense and dark red (a). As collagen deposition occurs, their color becomes gray (b). From Travis et al. Atlas of Nontumor Pathology: Volume 2: Non-Neoplastic Disorders of the Lower Respiratory Tract, American Registry of Pathology; 2002.
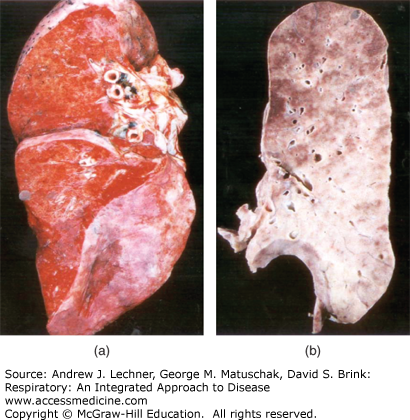
FIGURE 23.2
(a) Hyaline membranes (arrow) dominate the exudative phase of DAD, lining most airspaces in this image. (b) In the proliferative phase, fibroblasts infiltrate the hyaline membranes and expand the interstitium, thickening alveolar septa; type 2 pneumocyte hyperplasia (arrow) is common in the proliferative phase of DAD. (c) As fibroblasts synthesize collagen, DAD enters the fibrotic phase, with marked expansion of the interstitium by collagen; in this example, there is widespread squamous metaplasia (arrows). (c): From Travis et al. Atlas of Nontumor Pathology: Volume 2: Non-Neoplastic Disorders of the Lower Respiratory Tract, American Registry of Pathology; 2002.
Clinically, ARDS begins with dyspnea and tachypnea; early in its course, a chest radiograph may be normal. Subsequently, the patient develops cyanosis, hypoxemia, and respiratory failure, at which point a chest radiograph typically shows diffuse bilateral infiltrates. As hypoxemia becomes unresponsive to oxygen therapy, respiratory acidosis often develops. Of note, oxygen therapy can worsen the alveolar epithelial damage. ARDS can be complicated by secondary infection of the hyaline membranes and/or by death, the latter occurring in approximately 40% of cases in the United States.
Acute interstitial pneumonia is a rapidly progressive acute restrictive lung disease with a presentation similar to ARDS but without a known underlying etiology; an alternate name is idiopathic ALI-DAD. Morphologically, it closely resembles DAD and may be indistinguishable from DAD. Mortality rates in various studies have ranged from 33% to 74%. Survivors often experience complete or near complete recovery.
CHRONIC RESTRICTIVE LUNG DISEASES
The chronic restrictive lung diseases are also called diffuse interstitial lung diseases, because changes in the interstitium dominate the morphologic appearance, and diffuse infiltrative diseases, because chest radiographs show diffuse infiltrates. Chronic restrictive lung diseases are a heterogeneous group of disorders without uniform classification, without uniform terminology, and often without known etiology or pathogenesis. Nevertheless, they share many clinical and morphological features and, at end-stage, they may be indistinguishable from each other. Clinically, patients with chronic restrictive lung diseases have dyspnea, tachypnea, end-inspiratory crackles, and eventual cyanosis (Chap. 24). Later, these patients often develop secondary pulmonary hypertension (Chap. 26) and right heart failure with cor pulmonale. Pathogenetically, many of the chronic restrictive lung diseases begin with alveolitis, leading to distortion of alveolar structure and release of mediators that incite cell injury and induce fibrosis. Morphologically, many of the chronic restrictive lung diseases, particularly in later stages, are characterized by interstitial fibrosis. The end-stage of many of the chronic restrictive lung diseases is the classic honeycomb lung.
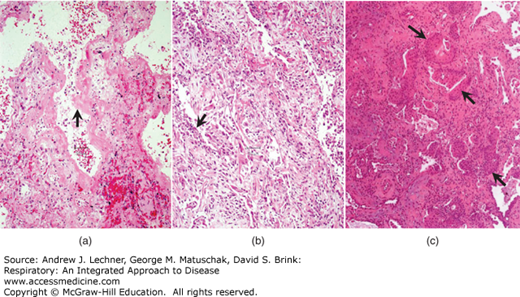
Idiopathic pulmonary fibrosis (IPF) is a poorly understood, idiopathic, nongranulomatous chronic restrictive lung disease that morphologically is characterized by diffuse interstitial fibrosis. Although many alternate names for the disease exist, cryptogenic fibrosing alveolitis is the one most frequently encountered. The pathogenesis of IPF is poorly understood but appears to involve repeated cycles of alveolitis (due to an unidentified agent) that are followed by wound healing with fibroblast proliferation. Grossly, lungs with well-developed IPF have a pleural surface with a cobblestone appearance due to wound contraction in interlobular septa. The cut surface of IPF lungs shows rubbery-to-firm, white patches in subpleural regions and in the interlobular septa (Fig. 23.3).
FIGURE 23.3
Usual interstitial pneumonia in idiopathic pulmonary fibrosis. Pale regions represent fibrosis with cystic change (honeycomb pattern) and are concentrated in the lower lobe and in the subpleural zone of the upper lobe (left of image). The darker parenchyma represents portions of lung with little or no fibrosis. From Travis et al. Atlas of Nontumor Pathology: Volume 2: Non-Neoplastic Disorders of the Lower Respiratory Tract, American Registry of Pathology; 2002.
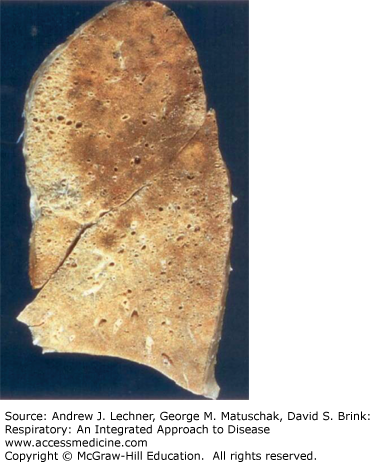
Histologically, the morphology of IPF is described as usual interstitial pneumonia (UIP) (Fig. 23.4). Although required for a diagnosis of IPF, UIP is not specific and can be seen in other diseases (eg, collagen vascular diseases, asbestosis; see below). UIP is characterized by regional and temporal heterogeneity where different lung foci show different stages of disease. In addition to interstitial fibrosis, magnified in subpleural zones and interlobular septa, UIP includes characteristic fibroblastic foci and typically shows prominent type 2 pneumocyte hyperplasia. End-stage UIP shows dilatated airspaces lined by cuboidal or low columnar epithelium separated by inflamed fibrous tissue. Patients with IPF typically present in the fifth to eighth decade with increasing dyspnea on exertion and dry cough, followed by hypoxemia, cyanosis, and digital clubbing. Progression of IPF is unpredictable, but the mean survival time is approximately three years. The only definitive therapy for IPF is lung transplantation.
FIGURE 23.4
Usual interstitial pneumonia in idiopathic pulmonary fibrosis. (a) Cystically dilatated airspaces are separated by fibrotic and thickened septa. (b) A characteristic finding in UIP is the fibroblastic focus (arrow). (b): From Travis et al. Atlas of Nontumor Pathology: Volume 2: Non-Neoplastic Disorders of the Lower Respiratory Tract, American Registry of Pathology; 2002.
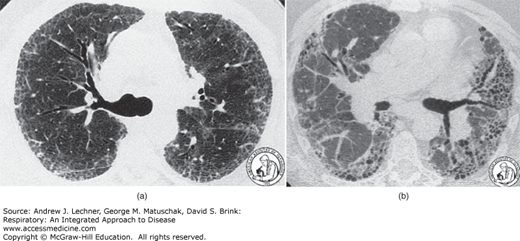
CLINICAL CORRELATION 23.1
By turning up the brightness on a computed tomogram, the resulting CT images of the lungs can demonstrate severity of interstitial disease. The settings used to image lung parenchyma are the lung windows and blur detail in surrounding soft tissue. Fig. 23.5(a) is high-resolution CT of UIP, with subpleural distribution of abnormalities. Fig. 23.5(b) is an HRCT of end-stage UIP with honeycombing that is more pronounced on the patient’s left side (right side of image).
FIGURE 23.5
See Clinical Correlation 23.1 for details. From Travis et al. Atlas of Nontumor Pathology: Volume 2: Non-Neoplastic Disorders of the Lower Respiratory Tract, American Registry of Pathology; 2002.
Nonspecific interstitial pneumonia (NSIP) is an idiopathic, nongranulomatous lung disease without the defining diagnostic features of better-characterized diseases. Histologically, NSIP can show a cellular pattern with mild to moderate expansion of the interstitium by lymphocytes and plasma cells with either a uniform or patchy distribution [Fig. 23.6(a)]. Alternatively, a fibrosing pattern with diffuse or patchy interstitial fibrosis is noted [Fig. 23.6(b)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


