OTHER CYANOTIC HEART DEFECTS IN THE NEONATE
Introduction
In the preceding chapters, common cyanotic congenital heart defects (CHDs), namely, transposition of the great arteries (TGA), tetralogy of Fallot (TOF), tricuspid atresia, total anomalous pulmonary venous connection (TAPVC), truncus arteriosus (TA), pulmonary atresia with intact ventricular septum (PA-IVS), Ebstein’s anomaly of the tricuspid valve and cardiac malpositions, including asplenia/polysplenia syndromes, were discussed. In this chapter, some of the other cyanotic CHDs, not discussed previously will be reviewed.
Double-Outlet Right Ventricle (DORV)
DORV is an uncommon CHD in which both the pulmonary artery (PA) and aorta (Ao) arise from the morphologic right ventricle (MRV)1–3 and accounts for less than 1% of all CHDs. Several definitions have been used in the past; the most commonly used definitions are: (1) both the Ao and PA come off of the right ventricle (RV) and neither great artery is in continuity with the atrioventricular (AV) valve and (2) one great vessel completely comes off of the RV and more than one-half of the other great vessel also arises from the RV. A large ventricular septal defect (VSD) is usually present and is the only exit for the left ventricle (LV).
This lesion is categorized based on the location of the VSD and presence of pulmonary stenosis (PS). In nearly 50% of the cases, the VSD is perimembraneous and subaortic in location and the left ventricular output is directed into the Ao. If there is no associated PS, the clinical presentation is similar to large VSD. If significant PS is present, the clinical features are those of TOF. In approximately 25% patients, the VSD is subpulmonary and the left ventricular output is largely directed into the PA. In these babies, the physiology is that of TGA and is generally referred to as Taussig–Bing anomaly. In the final 25% cases, the VSD is either doubly committed to both the Ao and PA or not committed to either great artery. The size and the position of the VSD and the severity of PS determine the pathophysiology and clinical presentation.
The VSD is typically large; however, it may be congenitally absent in very rare cases and causes left ventricular hypoplasia, or it may become smaller and spontaneously close after birth, resulting in severe left ventricular obstruction. Such VSDs are named physiologically advantageous VSDs.4–6 The PS is frequently valvar or subvalvar in location; pulmonary atresia may rarely be present. In patients with subpulmonary VSD, subaortic stenosis may be present and may be associated with aortic coarctation. The great vessels are abnormally related in most subjects: side-by-side in two-thirds, d-malposition in 25% and l-malposition in a small minority. Normal great artery relationship may be seen in a few patients, mainly those with subaortic VSD.
Clinical Features
The clinical features are principally dependent on the location of the VSD and existence of PS. If the VSD is subaortic and there is no PS, the clinical features are those of a large VSD. In the presence of significant PS, the clinical features are those of TOF. If the VSD is subpulmonary, the clinical presentation is similar to that of TGA.
Noninvasive Evaluation
Findings on chest roentgenogram largely depend upon the position of the VSD and level of right ventricular outflow narrowing. Cardiomegaly and increased pulmonary vascular markings are seen in babies with subaortic VSD and no PS, while mild or no cardiac enlargement and diminished pulmonary vascular markings are seen in the patients with PS. Subpulmonary VSD patients have features similar to TGA babies with cardiomegaly and increased pulmonary markings.
There is no characteristic pattern in the ECG of babies with DORV; however, right ventricular or biventricular hypertrophy is commonly seen. Severe left ventricular hypertrophy may be seen if the VSD becomes obstructive.
Echocardiogram is helpful in evaluation of the size and function of the ventricles, size and location of the VSD and relative positions of the great arteries (Figures 37.1 and 37.2). The degree of PS may be quantified with Doppler studies. Subaortic obstruction and coarctation of the aorta (CoA) should be scrutinized in babies with subpulmonary VSD.
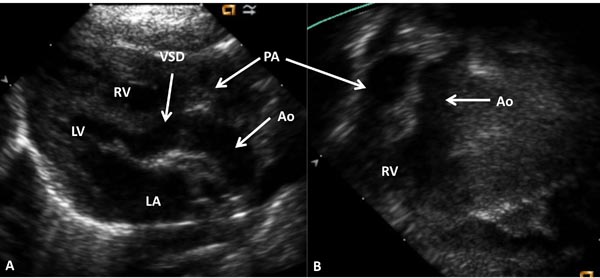
Figure 37.1. Selected video frames from precordial long-axis (A) and subcostal (B) views of a neonate with DORV and normally related great arteries demonstrating the origin of both the Ao and the PA from the RV. The VSD is shown. LA, left atrium; LV, left ventricle.
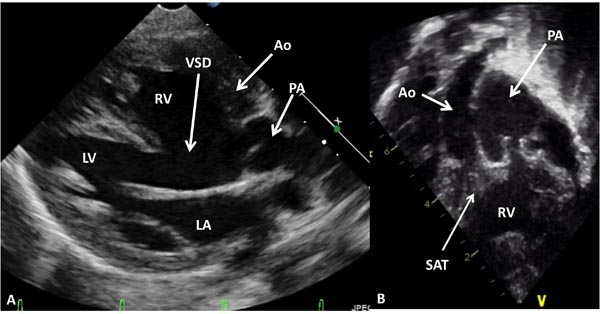
Figure 37.2. Selected video frames from precordial long-axis (A) and subcostal (B) views of another neonate with DORV and transposed great arteries demonstrating the origin of both the Ao and the PA from the RV. An extremely large VSD is also shown. LA, left atrium; LV, left ventricle; SAT, sub-aortic tissue.
Cardiac catheterization and selective cineangiography are usually not required because the echocardiographic studies clearly show the anatomy. However, in rare occasions, they may be needed to define the various issues alluded to.
Management
Initial management at presentation is similar to that described in the preceding chapters. In babies with subaortic VSD and no PS, surgical closure of the VSD to divert the left ventricular outflow into the Ao is the procedure of choice. However, such surgery is not usually necessary during the neonatal period. The naturally high pulmonary vascular resistance (PVR) at birth prevents rapid increase in pulmonary blood flow (PBF) and congestive heart failure (CHF). Indeed, delayed involution of pulmonary arterioles and late regression of PVR occurs in the presence of large interventricular communication (see Chapter 2) and it usually takes several weeks before marked increase in PBF and consequent CHF develop. We usually start these neonates on low doses of furosemide to prevent rapid onset of clinical heart failure. However, in some babies, rapid regression of PVR occurs, leading to CHF, and in such cases appropriate anticongestive measures should be initiated promptly, followed by surgery as mentioned above. If total surgical correction is not feasible, PA banding may be performed and surgical correction postponed to a later date.
In infants with subaortic VSD and PS, surgery is similar to that used for TOF including patch augmentation of the subpulmonary obstruction. Again, if the anatomy is not suitable for total surgical correction a modified Blalock–Taussig (BT) shunt7 may be performed with a plan for total correction later.
In babies with subpulmonary VSD (Taussig–Bing), arterial switch procedure8 along with VSD closure is optimal and can be performed safely at most institutions. Some of these babies may require balloon atrial septostomy9,10 because of poor mixing at atrial level while awaiting for surgical correction. CAo, if present, should also be addressed at the same time. In the presence of severe subaortic obstruction, Damus–Kaye–Stansel11 procedure may be needed. If there is associated PS, Rastelli type of surgery12 may be necessary.
Babies with uncommitted VSD are problematic and entail individualized plan to correct or palliate these infants.
Univentricular Hearts
The term univentricular heart has been used to describe any lesion with one functioning ventricle and includes double-inlet left ventricle (DILV), single ventricle, common ventricle, and univentricular atrioventricular (AV) connection.13 In such hearts, both AV valves may empty into the ventricle (DILV), one AV valve empties into the ventricle (with atresia of the other AV valve), or a single, common AV valve empties into the ventricle. DILV is the most common among the univentricular hearts and will be discussed. The management of the other entities is similar and will not be discussed separately.
The ventricle most commonly has left ventricular morphology in DILV, though right ventricular, mixed, indeterminate or undifferentiated morphologies may occur. The main ventricle is a morphological LV with an outlet chamber connected to it, which has a right ventricular morphology. The AV valves may be normal, hypoplastic, stenotic or even atretic. The great arteries are most frequently transposed and the Ao arises from the hypoplastic RV and the PA comes off of the main left ventricular chamber. l-Transposition occurs more often than d-transposition. The great arteries are normally related in less than 30% cases. DORV is also seen where both great vessels arise from the rudimentary RV. PS is present in two-thirds of the patients and such stenosis is present irrespective of great artery relationship. The stenosis may be at valvar or subvalvar level or the pulmonary valve (PV)/artery may be atretic. Subaortic obstruction may be present in patients with TGA and is due to the stenosis of the VSD or bulboventricular foramen (BVF). The latter group of patients often have associated aortic coarctation.
Clinical Features
The clinical presentation is principally dependent upon the degree of pulmonary outflow tract obstruction. In babies with PS, cyanosis and hypoxemia are the presenting symptoms. If the PS is severe, they may present early in the neonatal period. Increased ventricular impulse and a thrill at the left upper sternal border may be felt. On auscultation the second heart sound is single and a grade III to IV/VI long ejection systolic murmur at the left upper sternal border may be heard. Signs of CHF are notably absent.
Babies without PS usually present with symptoms of CHF within the first few weeks to months of life. In the neonatal period they may be detected because of abnormal prenatal obstetric ultrasound or because of a murmur on auscultation. There is mild, if any cyanosis because of increased PBF. The cardiac impulses are hyperdynamic. The cardiac sounds are usually normal with a nonspecific ejection systolic murmur (group I to II/VI) along the left sternal border and a mid-diastolic rumble at the apex. Signs of heart failure are not seen in the early neonatal period, but are present later.
Noninvasive Evaluation
The chest roentgenographic appearance varies depending upon the cardiac anatomy and amount of PBF. Mild cardiomegaly and decreased pulmonary vascular markings are seen in babies with diminished pulmonary flow. Moderate-to-severe enlargement of the heart and increased pulmonary vascular markings are noticed in babies with increased PBF. In babies with l-transposition, prominent and straight left heart border may be visualized.
ECG may show an abnormal initial QRS vector with a qR pattern in the right chest leads and/or Rs pattern in the left chest leads. Right, left or biventricular hypertrophy pattern may be present depending upon the anatomic type. However, none of the ECG features are pathagnomonic of DILV.
Echocardiogram is very helpful in making the diagnosis. The absence of the ventricular septum can be demonstrated (Figure 37.3). The AV connections and valves, ventriculoarterial connections and presence of PS can be determined (Figure 37.4). At times, cardiac catheterization and selective cineangiography may become necessary to define all the issues associated with DILV.
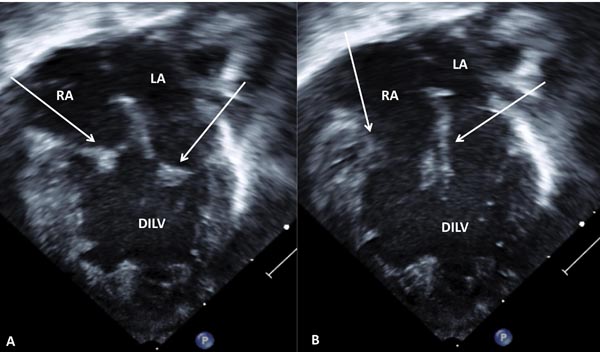
Figure 37.3. Selected video frames from apical 4-chamber views of a neonate with DILV with closed (A) and open (B) AV valves (arrows). The outlet chamber is not visualized in this view (see Figure 37.4). LA, left atrium; RA, right atrium.
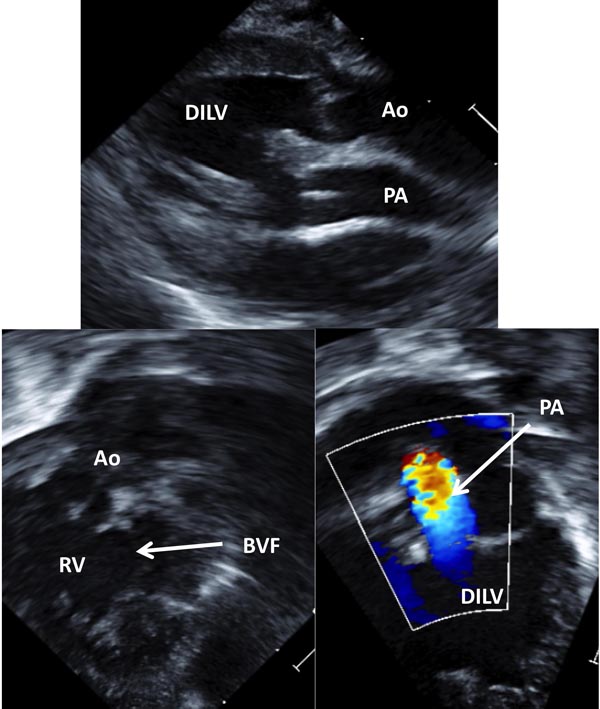
Figure 37.4. Selected video frames from precordial long-axis (A) and modified apical (B and C) views of the same neonate shown in Figure 37.3 demonstrating connection of the RV (outlet chamber) with the main chamber by a BVF. This chamber gives rise to the Ao as seen in B. The origin of the PA from the main chamber, DILV is shown in C. Note turbulent flow in the PA (C) suggesting stenosis.
Management
After the description by Fontan and Kreutzer of physiologically corrective operation for tricuspid atresia,14,15 the concept was widely adapted and the procedure extended to treat other cardiac defects including DILV. Staged total cavopulmonary connection is the Fontan procedure of choice at the present time; procedures to normalize PBF in the neonatal period, bidirectional Glenn at about 6 month of age and Fontan conversion with an extracardiac conduit and fenestration 1 year or 2 years after Glenn.
Neonates and young infants with PS resulting in moderate-to-severe arterial desaturation (O2 saturations ≤70%) are candidates for modified BT shunt.7 If the pulmonary outflow tract obstruction is predominantly at valvar level, balloon pulmonary valvuloplasty may be used to augment PBF.16–18 In babies with pulmonary atresia, intravenous administration of prostaglandin E1 (PGE1) followed by BT shunt (see Chapter 29) should be undertaken.
In infants with pulmonary plethora, the CHF should be aggressively treated followed by early PA banding. Successful banding should not only provide good control of CHF, but also reduce the pressure in PA so that the infants may become good risk candidates for eventual Fontan conversion. If there is aortic coarctation, it should be relieved promptly. Development of subaortic obstruction following banding has been demonstrated, but the author’s view is that progression to subaortic stenosis in likely to be time-related natural history of the disease rather than a direct relationship to banding.19 Should subaortic obstruction develop, bypassing the obstruction by Damus–Kaye–Stansel12 or resection of subaortic obstruction should be incorporated into the treatment plan.
The neonatal palliation should be followed by bidirectional Glenn and Fontan as mentioned above.
Interrupted Aortic Arch (IAA)
IAA is defined as a complete lack of luminal communication between the ascending aorta (AAo) and descending aorta (DAo); this is in contrast to aortic coarctation in which there is usually a luminal connection.20 This lesion comprises less than 1% of all CHD. Three types of IAA are described21: types A, B and C. Each type may be subgrouped depending on the origin and/or course of the subclavian arteries. In type A, the arch interruption is distal to left subclavian artery; in type B, the arch discontinuity is between the left common carotid artery and the left subclavian artery and in type C, the arch interruption is between the right innominate artery and the left common carotid artery. Type B interruption (between the left common carotid and left subclavian artery) is the most frequent, comprising of 52% of all cases.22 Type A interruption (distal to left subclavian artery) is second most common, accounting for 44% of cases while type C interruption (between the right innominate artery and the left common carotid artery) is least common, with only 4% of cases.22 The IAA occurs both with left and right aortic arches.
IAA is generally associated with other lesions. Patent ductus arteriosus (PDA) is most common and establishes continuity between the main pulmonary artery (MPA) and the DAo. VSD is present in nearly 80% of type B IAA cases while it is seen in 50% of babies with type A interruption. Valvar and subvalvar aortic stenosis, TA, DORV of Taussig–Bing type, single ventricle (DILV), TGA, and aortopulmonary septal defects are also seen in association with IAA. A strong association with DiGeorge’s syndrome (chromosome 22q11 microdeletion) has been observed, particularly with type B interruption.
Clinical Features
Because of patency of the ductus arteriosus (DA) and high PVR, the babies are usually asymptomatic at birth. As the ductus begins to constrict and/or PVR drops the infants become symptomatic. If the ductus closes completely, the babies will have symptoms of cardiovascular collapse and shock including tachycardia, tachypnea, poor pulses, and ashen color. With partially constricted ductus, blood pressure difference between upper and lower extremities maybe recorded. There is no blood pressure differential if there is an unrestricted PDA. Although the lower half of the body is perfused by the RV via the PDA, deferential cyanosis (upper extremity pink, lower extremity cyanotic) is not usually seen because of shunting of oxygenated blood across the VSD into the RV and PA. A murmur of VSD may be heard in babies without ductal closure. High degree of suspicion is necessary to entertain a diagnosis of IAA.
Noninvasive Evaluation
Chest X-ray shows cardiomegaly and increased pulmonary vascular markings. ECG may reveal right, left or biventricular hypertrophy. The corrected QTc interval may be prolonged secondary to hypocalcaemia in babies with DiGeorge’s syndrome. Two-dimensional (2D) echocardiographic studies23 usually confirm (Figure 37.5) the diagnosis. Magnetic resonance imaging studies and angiography are not usually necessary to establish a diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


