Nonischemic Cardiomyopathies
Carmela D. Tan
Norman B. Ratliff
James B. Young
E. Rene Rodriguez
Overview
Cardiomyopathies are defined by the World Health Organization as diseases of the myocardium associated with cardiac dysfunction. They are classified to reflect the recent insight gained into the pathogenesis of the heart muscle disorders. Cardiomyopathies are categorized into dilated, restrictive, hypertrophic, and unclassified based on the predominant pathophysiologic characteristics. Figure 89.1 shows that there is predominant remodeling of the left ventricle that distinguishes these categories. A new category has been added to include right ventricular abnormalities. The disorders that are associated with systemic or certain cardiac diseases are called specific heart muscle diseases and include ischemic cardiomyopathy, valvular, hypertensive, inflammatory, metabolic, peripartal, general systemic disease, muscular dystrophies, neuromuscular disorders, and toxic and hypersensitivity reactions. The unclassified cardiomyopathy category includes disorders such as fibroelastosis, noncompacted myocardium, and systolic dysfunction with minimal dilation. These diseases either have features that overlap the other classifications or do not readily fit any category. The identification of the etiology of any patient who presents with cardiomyopathy is very important, because specific treatment may reverse cardiac dysfunction. Endomyocardial biopsy is useful to diagnose specific diseases, as part of a clinical trial, to differentiate between restrictive and constrictive disorders, and to monitor anthracycline toxicity. Routine biopsies for idiopathic dilated cardiomyopathies (DCM) are helpful to establish specific diagnosis. Future directions in the diagnosis and management of cardiomyopathies include elucidating genetic and molecular mechanisms of their pathogenesis and the role of these mechanisms in heart failure.
Historical Perspective
In 1891, one of the earliest descriptions of heart muscle diseases appeared (1). Unfortunately, little more about these “obscure disorders” was gleaned in the six decades that followed. In 1957, Brigden highlighted the ignorance and confusion that existed regarding “noncoronary cardiomyopathies,” their terminology, and clinicians’ reluctance to diagnose them (2). Since then, medical science has amassed considerable insight regarding the pathophysiology and treatment of these prevalent disorders. Although remarkable progress has been made, there is considerable need for further investigation.
To understand more fully the current nomenclature relating to myocardial failure, it is helpful to review the original working terminology of heart muscle disorders. In 1980, the World Health Organization and International Society and Federation of Cardiology task force met in Paris to clarify this area. They defined the cardiomyopathies as “heart muscle diseases of unknown cause” and divided them into three broad classifications based on characteristic anatomic findings of the heart: dilation, hypertrophy, and restriction (3). A fourth group, “unclassified cardiomyopathies,” was also listed to include cases that did not easily fit into one of the other three general categories. Unclassified cardiomyopathies were sometimes referred to as latent cardiomyopathies. Completely separate from the cardiomyopathies were specific heart muscle diseases, characterized
as diseases of known cause or associated with specific disorders of other systems. They were further divided into infective, metabolic, general system diseases, heredofamilial, hypersensitivity, and toxic reactions categories. Myocardial dysfunction as a result of systemic or pulmonary hypertension, coronary artery disease, valvular heart disease, or congenital anomalies was excluded.
as diseases of known cause or associated with specific disorders of other systems. They were further divided into infective, metabolic, general system diseases, heredofamilial, hypersensitivity, and toxic reactions categories. Myocardial dysfunction as a result of systemic or pulmonary hypertension, coronary artery disease, valvular heart disease, or congenital anomalies was excluded.
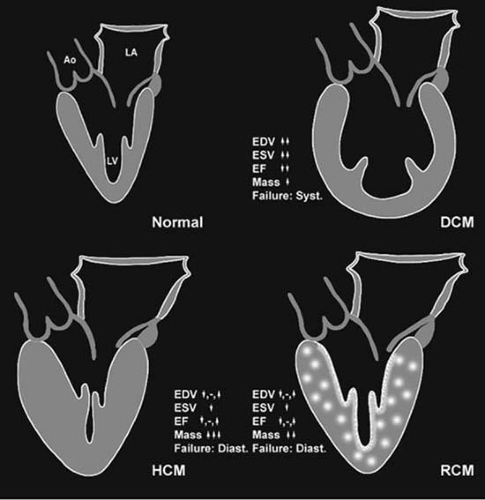 FIGURE 89.1. Morphologic features of the main types of cardiomyopathies. The normal geometry of the LV is shown. In comparison, there is enlargement and dilatation of the LV in DCM. In HCM, there is marked thickening of the LV wall, often asymmetric, with the septum being even thicker than the free wall of the LV. In restrictive cardiomyopathy (RCM), the ventricular wall may be normal, hypertrophic, or slightly dilated, but the main feature is that the restriction to diastolic compliance of the ventricle is intrinsic to the ventricle. This occurs by involvement of the endocardium or more commonly by infiltration of the interstitial space with excess collagen, amyloid, or granulomata and their ensuing fibrosis. Abbreviations: EDV, end-diastolic volume; ESV, end-systolic volume; EF, ejection fraction; Mass, ventricular mass; Failure, mode of failure, systolic versus diastolic. |
Etymologically, cardiomyopathy refers to a disease that arises from the heart itself (4). When clinical heart failure syndromes are diagnosed, it is the functional integrity of the circulatory network—including the heart—that is important for characterization of heart failure. Obviously, all cardiomyopathies have some etiologic factor. Many times, the specific reason for the cardiac malfunction and subsequent remodeling cannot be precisely characterized. The term idiopathic is used to describe this clinical situation. It is not a specific disease entity, but likely represents a wide spectrum of difficulties and is a term that largely reflects our ignorance regarding the etiology of heart failure in many given instances. As more became known about the individual disorders, it was apparent that new definitions and classifications were needed, and in 1996, The World Health Organizations released a revised categorization (5) (Table 89.1). Cardiomyopathies were generally more simply defined as diseases of the myocardium associated with cardiac dysfunction. As in the previous scheme, dilated, hypertrophic, restrictive, and unclassified divisions remained. However, arrhythmogenic right ventricular cardiomyopathy (ARVC) was added. Further, specific heart muscle diseases were referred to as specific cardiomyopathies that included myocardial dysfunction associated with distinct cardiac or systemic disorders.
Dilated Cardiomyopathy
DCM is characterized by ventricular remodeling that produces chamber dilation, with normal or decreased wall thickness, and diminution in systolic function (Fig. 89.2). The impairment of systolic function can involve the left, right, or both ventricles, with the ejection fraction traditionally being defined as less than 40% (6). Although the total mass of the heart is increased, the ratio of left ventricular (LV) volume to ventricular wall thickness is much greater than for patients with hypertrophic cardiomyopathy (HCM) or diastolic dysfunction owing to hypertensive heart disease (7). This eccentric hypertrophy provides a unifying concept for many different disease processes, ultimately resulting in dilation of the heart. It is important to note that remodeling of this sort is caused by diseases that are, for the most part, distinct from those causing hypertrophic and restrictive cardiomyopathy, problems that generally do not result in chamber dilation. Myocardial remodeling is associated with altered hemodynamics characterized by diminished ejection fraction, reduced stroke volume, increased chamber volumes, and, therefore, increased chamber pressures. This triggers the multiple neurohormonal aberrations that are characteristic of the heart failure milieu.
Often, if the toxic or injurious agent can be identified and removed, further detrimental chamber remodeling can be
blocked and systolic function returned to a more normal state. However, DCMs generally pass through an early and late evolutionary period. The stages parallel pathophysiologic events triggered by whatever myocardial insult initiated the heart failure process in the first place. At some point, injury produces myocyte and myocardial dysfunction that antedates clinically apparent disease. As progression occurs and decompensation develops, symptoms begin to appear that are characteristic of heart failure in general. Disease-specific cardiac and extracardiac signs and symptoms may provide insight to the etiology of the heart failure and cardiomyopathy. If the process is not interdicted, later stages will appear and manifest with profound eccentric hypertrophy and, ultimately, marked reduction in systemic flows accompanying elevation of intracardiac pressures.
blocked and systolic function returned to a more normal state. However, DCMs generally pass through an early and late evolutionary period. The stages parallel pathophysiologic events triggered by whatever myocardial insult initiated the heart failure process in the first place. At some point, injury produces myocyte and myocardial dysfunction that antedates clinically apparent disease. As progression occurs and decompensation develops, symptoms begin to appear that are characteristic of heart failure in general. Disease-specific cardiac and extracardiac signs and symptoms may provide insight to the etiology of the heart failure and cardiomyopathy. If the process is not interdicted, later stages will appear and manifest with profound eccentric hypertrophy and, ultimately, marked reduction in systemic flows accompanying elevation of intracardiac pressures.
TABLE 89.1 World Health Organization Classification of Cardiomyopathies | |
|---|---|
|
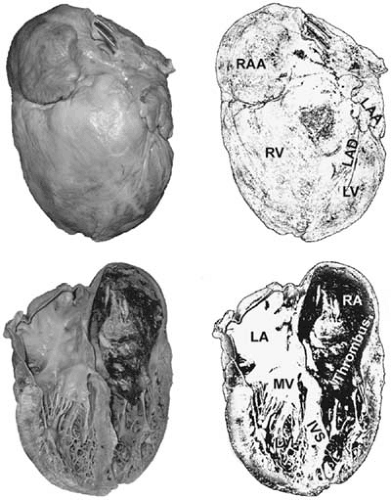 FIGURE 89.2. Dilated cardiomyopathy. There is marked global enlargement of the heart. In this case, there is enlargement of the left- and right-sided cavities as the result of severe LV failure. There is also a thrombus in the right atrium. Abbreviations: IVS, interventricular septum; LA, left atrium; LAA, left atrial appendage; LAD, left anterior descending coronary artery; LV, left ventricle; MV, mitral valve; RA, right atrium; RAA, right atrial appendage; RV, right ventricle. |
DCM was previously referred to as congestive cardiomyopathy; however, this term is no longer applied because congestion is not consistently present, particularly in the milder and earlier stages or after therapy. DCM is the most common of the cardiomyopathies, comprising more than 90% of all cases that are referred to specialized centers (8), and represents the end result of more than 50 distinct diseases (9,10). When diagnosing DCM, it is important to review the major etiologic and pathophysiologic classes responsible for this disorder (see Table 89.1). By and large, DCMs can be familial or genetic in origin; secondary to infection or inflammation, toxic substance exposure, metabolic derangements; or idiopathic.
Some progress has been made in characterizing the molecular genetics of DCM (11). Perhaps as many as 20% of DCM cases are inherited or familial, with a significant percentage of the remaining being acquired as suggested. Inherited forms of DCM may have autosomal-dominant, autosomal-recessive, X-linked, or mitochondrial transmission. Mutations have been found in gene encoding for proteins that fall in four general classes according to their function in the cardiac myocyte: (a) force generation and propagation (sarcomere components, M-band components, Z-disc components, cytoskeletal components); (b) energy production and regulation (mitochondrial mutations, glycogen metabolism); (c) calcium cycling; and (d) transcriptional regulators (Nkx2.5, NFAT3, GATA4, HDACs, MEF2, Hop, PPARs) (12).
Idiopathic Dilated Cardiomyopathy
Background and Prevalence
Idiopathic DCM is a primary global myocardial disorder of unknown cause. Because it is the pathophysiologic model for other specific DCMs, this section focuses on the idiopathic type, with distinguishing characteristics of other specific disorders discussed subsequently. Idiopathic DCM alone accounts for 36 cases per 100,000 population, 10,000 deaths in the United States per year, and approximately 25% of all cases of cardiomyopathy.
Pathology
As shown in Figure 89.1, there are anatomic differences in the cardiomyopathies, emphasizing that DCM has marked enlargement of LV end-diastolic and end-systolic volumes, usually with profound reduction of the ejection fraction. This is in contrast to restrictive and hypertrophic cardiomyopathies, which have more normal end-diastolic volumes and, sometimes, supernormal ejection fractions. In DCM, the mitral and tricuspid annuli are frequently enlarged from ventricular dilation, with resultant valvular regurgitation. By definition, idiopathic DCM is not caused by coronary artery disease, and therefore the coronary arterial tree is usually normal. It is possible to have some plaque formation; however, the degree of systolic impairment is out of proportion to atherosclerotic involvement. The histology is usually nonspecific, but certain features are present. Under the light microscope, the myocytes are commonly quite hypertrophied. The nuclei may be very large with bizarre shapes. The areas of interstitial fibrosis are frequently underappreciated. Occasionally, small clusters of lymphocytes in the interstitium or in perivascular location can be present. By electron microscopy, the mitochondria may be abnormal, the T tubules dilated, and lipid vacuoles present.
Clinical Profile and Management
Chapters 86 and 87 discuss in detail the treatment of acute and chronic heart failure. Management options include supportive therapy (diet, sodium, toxin and fluid restriction, exercise), medications (diuretics, digoxin, angiotensin-converting enzyme [ACE] inhibitors, angiotensin II receptor blockers, inotropes, anticoagulants, antiarrhythmics, and β-blockers), ventricular assist devices, and novel surgical procedures (cardiomyoplasty, mitral and tricuspid valve repair, partial left ventriculectomy, and cardiac transplantation).
Hypertrophic Cardiomyopathy
Background and Prevalence
HCM is addressed in detail in Chapter 29. Since the modern description by Teare in 1958 (13), HCM has been known by a confusing array of names that largely reflect its clinical heterogeneity, relatively uncommon occurrence in cardiologic practice, and the skewed experience of early investigators. This problem in nomenclature has been an obstacle to the general recognition of the disease within the medical and nonmedical communities. Hypertrophic cardiomyopathy (or HCM) is now widely accepted as the preferred term (14) because it describes the overall disease spectrum without introducing misleading inferences that LV outflow tract obstruction is an invariable feature of the disease, such as is the case with hypertrophic obstructive cardiomyopathy, muscular subaortic stenosis, or idiopathic hypertrophic subaortic stenosis. Indeed, most patients with HCM do not demonstrate outflow obstruction under resting (basal) conditions, although many may develop dynamic subaortic gradients of varying magnitude with provocative maneuvers or agents (14). The contemporary definition of HCM focuses on the fact that this disorder is characterized by disproportionate and remarkable left, and sometimes right, ventricular hypertrophy (14,15). Typically, the septum is more involved than the free LV wall, although on occasion the hypertrophy can be completely concentric in nature (Fig. 89.3). HCM, which may develop at any age, is associated with marked LV volume reduction, and systolic LV outflow tract gradients are common. Up to 70% of cases have a familial pattern of occurrence, with autosomal-dominant inheritance. Clinically, the diagnosis of HCM is most reliably made by two-dimensional echocardiography showing left ventricular
hypertrophy (LVH) with asymmetric distribution. This LVH is associated with a nondilated and hyperdynamic chamber, which often shows systolic LV cavity obliteration in the absence of another cardiac or systemic disease such as hypertension or aortic stenosis. Although the usual clinical diagnostic criteria for HCM is a maximal LV wall thickness greater than or equal to 15 mm, genotype–phenotype correlations have shown that virtually any wall thickness is compatible with the presence of a HCM mutant gene (14,15).
hypertrophy (LVH) with asymmetric distribution. This LVH is associated with a nondilated and hyperdynamic chamber, which often shows systolic LV cavity obliteration in the absence of another cardiac or systemic disease such as hypertension or aortic stenosis. Although the usual clinical diagnostic criteria for HCM is a maximal LV wall thickness greater than or equal to 15 mm, genotype–phenotype correlations have shown that virtually any wall thickness is compatible with the presence of a HCM mutant gene (14,15).
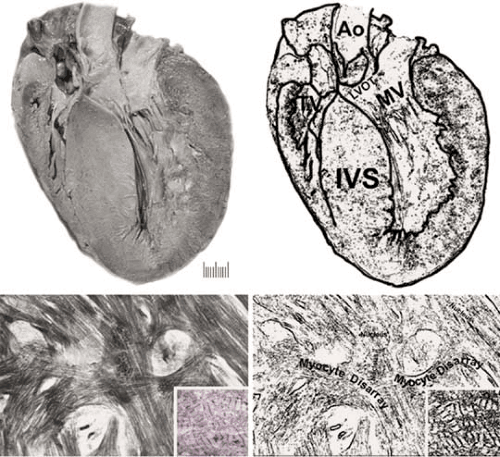 FIGURE 89.3. Hypertrophic cardiomyopathy. The gross image shows a coronal section of a heart with marked asymmetric septal hypertrophy. The interventricular septum is 2.5 times thicker than the LV free wall. The anterior leaflet of the mitral valve is in very close apposition to the LV outflow tract. The bottom panel is a light micrograph showing marked myocyte disarray, where the individual myocytes no longer have a parallel orientation with respect to each other. Instead, they have a “storiform” array of the myofibrils. The inset shows that the myofibrils themselves and the sarcomeres are disarrayed when examined by transmission electron microscopy. Abbreviations: Ao, aorta; IVS, interventricular septum; LVOT, left ventricular outflow tract; MV, mitral valve. |
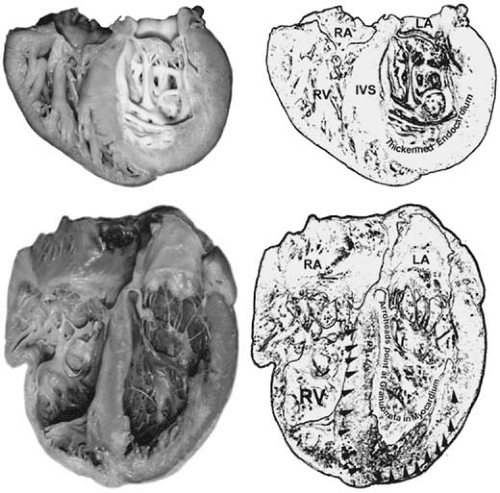 FIGURE 89.4. Restrictive/infiltrative cardiomyopathy. The gross image on top shows a heart with marked endocardial fibroelastosis which prevents diastolic compliance of the left ventricle. Note the markedly thick (white) endocardium. The heart in the lower part of the panel is a heart with patchy pale/white areas involving the interventricular septum from the base to the apex and continuing into the free wall of the left ventricle owing to infiltration by noncaseating granulomata in a case of sarcoidosis. Abbreviations: IVS, interventricular septum; LA, left atrium; RA, right atrium; RV, right ventricle. |
Pathology
The morphologic hallmark of HCM is asymmetric septal hypertrophy (see Figure 89.3) with myocyte and myofibril disarray (16). Some reports have emphasized that variable degrees of myocyte ultrastructural, myofibrillar, and muscle bundle disorganization can be detected, often with substantial degrees of interstitial fibrosis (16). Mutations in sarcomeric protein encoding genes have been described in HCM, but other conditions can mimic the phenotype such as Noonan syndrome, Friedreich ataxia, Fabry disease, and Hunter and Hurler syndromes.
Recently, a proposal for screening patients with familial HCM suggests the use of echocardiography from the age of 12 until full growth and maturation is achieved (18–21 years). If echocardiographic abnormalities are not demonstrated, then a strong reassurance to the family can be made regarding the absence of cardiomyopathy (17). The medical therapy of HCM is discussed in Chapter 29. Other modalities of therapy for patients with outflow tract obstruction include ethanol ablation (via catheterization) and surgical resection of the outflow tract hypertrophied muscle (myectomy). This last procedure is better in terms of outcome (18).
Restrictive Cardiomyopathy
The restrictive or infiltrative cardiomyopathies (Fig. 89.4), sometimes referred to as nondilated, nonhypertrophic cardiomyopathies, are described in Chapter 27. Generally, this is the least common of the major cardiomyopathies and is characterized by a normal or only slightly enlarged heart, decreased diastolic volumes, and, early in the disease, normal systolic function (19). The main pathologic disorder is one of impaired diastolic functioning, resulting from decreased ventricular compliance and filling. This dysfunction may result from a process that involves, primarily, the endocardium, myocardium, or both. The most commonly encountered myocardial disorder is amyloid heart disease (Fig. 89.5) and other specific myocardial forms include metabolic storage disorders such as Fabry disease and hemochromatosis, sarcoid heart disease, radiation fibrosis, and various tumor infiltrations, many of which are also addressed in the section on specific cardiomyopathies. The endocardial disorders, less common in the United States, include entities such as endomyocardial fibrosis, Löeffler endocardial fibrosis and, even more rarely, endocardial fibroelastosis. Endocardial fibroelastosis is typically placed in the unclassified cardiomyopathy category. Restrictive cardiomyopathy should be considered when patients present with signs and symptoms of myocardial failure but normal cardiac size. Frequently, right-sided findings of increased jugular venous pressure, ascites, and peripheral edema predominate, although left-sided failure does occur. The differential diagnosis includes constrictive pericarditis, which is a more readily treatable condition. Echocardiography and right heart catheterization may help to differentiate these disorders. Endomyocardial biopsy should also be considered if other historical and physical clues to the diagnosis are not present. The treatment of
restrictive cardiomyopathy is also reviewed in Chapter 27 and generally remains quite unsatisfactory.
restrictive cardiomyopathy is also reviewed in Chapter 27 and generally remains quite unsatisfactory.
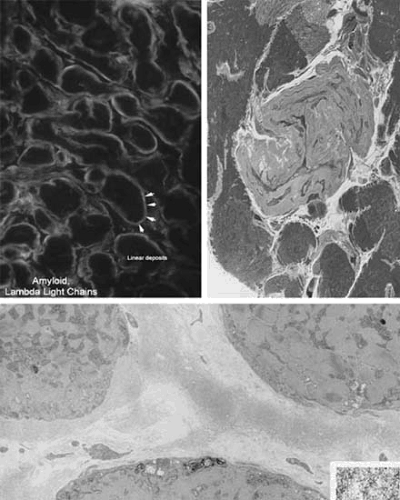 FIGURE 89.5. Amyloidosis of the heart. The left upper panel shows a thioflavin-S stain showing thick, linear, ring-like deposits of amyloid (white arrows) surrounding individual myocytes. In this particular example, the protein responsible for the formation of these amyloid deposits is the λ light chain. The image on the upper right side shows a “nodular deposit” of amyloid. The bottom image shows typical electron microscopic view of perimyocytic deposits of 10-nm thick amyloid fibrils in the interstitium of the heart. The lower inset shows a close up of the amyloid fibrils. |
Endomyocardial Fibrosis
Endomyocardial fibrosis, also known as tropical eosinophilic endomyocardial fibrosis, is common in South and Central America and tropical and subtropical Africa. Surprisingly, it accounts for 15% to 25% of the cardiac deaths in equatorial Africa (20). Endomyocardial fibrosis is characterized by thickening and scarring of the endocardium, which can be so extensive that the ventricular cavity is obliterated. Thrombi may overlie these lesions, increasing the risk of thromboembolism. Frequently, the subendocardial myocardium is also affected. In 50% of cases, the left and the right ventricles are involved, whereas another 40% of cases have lesions isolated to the left ventricle. The mitral and tricuspid valves may become fibrotic as well, leading to valvular regurgitation. Many believe that this disease is a form of, if not the same as, Löeffler endocardial fibrosis, perhaps at a different stage (20,21). Although the overall prognosis is poor, palliative treatment with surgical endocardial resection and valvular repair/replacement may alleviate some symptoms.
Löeffler Endocardial Fibrosis
Löeffler endocardial fibrosis is also associated with eosinophilia, giving it its other names, eosinophilic cardiomyopathy and nontropical eosinophilic endomyocardial fibrosis. The severity of cardiac dysfunction seems to parallel the degree and duration of the eosinophilia, leading to the belief that the eosinophil itself is responsible for the dysfunction. The contents of the intracytoplasmic granules of the eosinophils can cause direct myocardial necrosis. It has also been observed that the eosinophils bind immunoglobulin G and increase peroxidase, which can have further direct toxic effects. The cardiac manifestations of this eosinophilic disorder are similar, if not identical to, those produced by eosinophilic leukemia and the eosinophilia–myalgia syndrome, which has been associated with tryptophan. Treatment with corticosteroids and cytotoxic agents (hydroxyurea) can improve symptoms and survival (22).
Right Ventricular Cardiomyopathy—Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia
Background and Prevalence
ARVC, also commonly known as arrhythmogenic right ventricular dysplasia (ARVD), is a rare disorder that occurs in adolescence and young adulthood. It has a male : female ratio of approximately 2 to 3 : 1 (23). The true incidence of ARVC
remains unknown, partly because of difficulties with its diagnosis, as described below.
remains unknown, partly because of difficulties with its diagnosis, as described below.
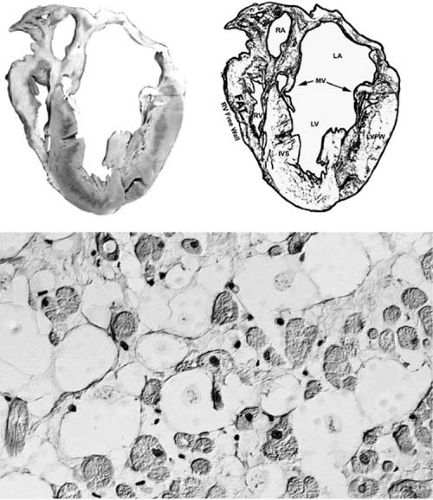 FIGURE 89.6. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). In this gross specimen, the right ventricular free wall has been replaced by adipose tissue, which is noticeable from the atrioventricular groove, just below the right atrium, all the way to the junction of the right ventricular free wall to the interventricular septum. The bottom panel shows a light micrograph of the adipose tissue infiltration in the right ventricular wall. Abbreviations: LA, left atrium; LV, left ventricle; MV, mitral valve; RA, right atrium. |
Pathology
The abnormalities of ARVC affect primarily the right ventricle, although in rare instances, the left ventricle is involved (24,25). Grossly, the ventricle is mildly dilated and thinned (Fig. 89.6). The characteristic feature is the segmental, progressive, noninflammatory loss of myocytes and their replacement by adipose tissue and fibrosis (26). This is in contrast to the nonfibrotic adipose tissue infiltration that normally occurs in the right ventricle and increases as a function of age (27). In ARVC/D, the fibroadipose infiltration is transmural in nature and can be confirmed either at autopsy or with surgical biopsy. Endomyocardial biopsies are sometimes helpful, but have limitations (28). Despite this, finding extensive fibrofatty replacement of myocytes in the right ventricle is concerning. But when interpreted by an experienced team including cardiologists, pathologists, and radiologists, the diagnosis of ARVD can be accurately established (29). Cinemagnetic resonance imaging has the potential to aid diagnosis by being able to differentiate normal myocardium from adipose tissue, assess morphologic alterations, and detect areas of regional and global ventricular dysfunction (30). In fact, a good correlation between imaging and tissue characterization of ARVD has been demonstrated (31).
Genetic studies from several sources have identified at least 7 loci (ARVD1-ARVD7) by linkage analysis. Furthermore, mutations in three desmosomal proteins (desmoplakin, plakoglobin with specific phenotype or Naxos disease [palm plantar keratoderma and wooly hair], and plakophilin) (32,33) have been associated with ARVC. Also, mutations in the cardiac ryanodine receptor (hRYR2), are associated with effort-induced polymorphic ventricular tachycardia and juvenile sudden death (ARVD2) (33). Last, mutations in the transforming growth factor-β3 have been associated with an autosomal dominant inheritance (ARVD1).
Clinical Profile and Management
Because of the difficulties and risks of obtaining tissue to confirm the diagnosis of ARVC, as well as inaccuracies in assessing right ventricular structure and function with many of the noninvasive tests, a task force from the European Society of Cardiology and the International Society and Federation of Cardiology met and released criteria to assist in this process. The diagnosis can be made by meeting any of the following: two major criteria, one major plus two minor, or four minor criteria (Table 89.2) (34). A recent report of electrocardiographic (ECG) changes evaluated over time shows usefulness of the ECG in assessing progression of ARVC/D (35).
To assess the natural history of ARVC/D, a recent study of 130 patients showed a cardiovascular mortality of 16% (n = 24). Risk stratification showed that the most ominous risk factors presence of right or left ventricular dysfunction and ventricular tachycardia. However, when cause of death in these 24 patients was analyzed, 59% of the patients died of heart failure and 29% of sudden death (36).
UHL Anomaly
This entity shows marked thinning of the right ventricular wall with virtual apposition of the epicardium to the endocardium
without myocardium in between (37). This anomaly has been compared to ARVC/D. However, it is distinct in that it occurs in infancy and early childhood, is incessant, and results in complete destruction of the right ventricle. Furthermore, it does not show fatty infiltration of the wall (38).
without myocardium in between (37). This anomaly has been compared to ARVC/D. However, it is distinct in that it occurs in infancy and early childhood, is incessant, and results in complete destruction of the right ventricle. Furthermore, it does not show fatty infiltration of the wall (38).
TABLE 89.2 Criteria for Diagnosis of ARVD/C | ||
|---|---|---|
|
Unclassified Cardiomyopathies
Endocardial Fibroelastosis
Endocardial fibroelastosis is an abnormal thickening of the endocardium of the left ventricle and aortic and mitral valves. It occurs in infancy and early childhood. The fibrosis leads to decreased compliance of the myocardium and diastolic dysfunction. Typically, a DCM occurs frequently with significant mitral regurgitation, but a restrictive cardiomyopathy (see Fig. 89.4) can also occur. A genetic defect has not been identified; autosomal-recessive, autosomal-dominant, and X-linked recessive patterns of inheritance have been reported. The X-linked forms demonstrate mitochondrial abnormalities that are similar to those of Barth syndrome. Endocardial fibroelastosis has also been associated with tissue carnitine deficiency (39), and there is evidence that it may be related to a defect in the plasma membrane carnitine transporter (40). The ECG frequently shows features of LVH, with occasional pseudoinfarction patterns and atrioventricular block. The echocardiogram is remarkable for dense echoes along the LV endocardium. Pathologic examination shows a uniform thickening of the encodardium of the left ventricle, which is composed of a mixture of collagen and elastic fibers.
Noncompacted Myocardium
Left ventricular noncompaction (LVNC) is a rare form of cardiomyopathy believed to be a result of an arrest in cardiac
development (Fig. 89.7). It affects adults and children. In adults, it is often limited to patients without associated cardiac defects, and is thus called isolated LVNC. In children, it is associated with other heart defects in up to 14% of patients. In children, there are two phenotypes: (a) progressive systolic dysfunction that results in early death; and (b) an undulating type, with periods of deterioration and recovery that allow survival into adulthood. Other terms for persistent noncompacted myocardium include spongy myocardium and persistence of myocardial sinusoids; however, the intramyocardial vessels that communicate with the cardiac chambers are invaginations rather than sinusoids (41,42). There are associations of LVNC with mutation in the gene G4.5, which encodes tafazzin. This gene has also been associated with other myopathies and with Barth syndrome (43,44).
development (Fig. 89.7). It affects adults and children. In adults, it is often limited to patients without associated cardiac defects, and is thus called isolated LVNC. In children, it is associated with other heart defects in up to 14% of patients. In children, there are two phenotypes: (a) progressive systolic dysfunction that results in early death; and (b) an undulating type, with periods of deterioration and recovery that allow survival into adulthood. Other terms for persistent noncompacted myocardium include spongy myocardium and persistence of myocardial sinusoids; however, the intramyocardial vessels that communicate with the cardiac chambers are invaginations rather than sinusoids (41,42). There are associations of LVNC with mutation in the gene G4.5, which encodes tafazzin. This gene has also been associated with other myopathies and with Barth syndrome (43,44).
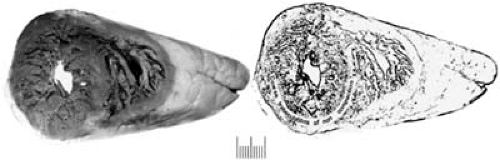 FIGURE 89.7. Left ventricular noncompaction. Note the coarse trabeculations present throughout the LV wall. These trabeculations closely resemble the trabeculation pattern of the right ventricle. |
Mitochondrial Cardiomyopathy
Mitochondrial cardiomyopathy is defined as ventricular dysfunction caused by mitochondrial DNA mutations, which are maternally inherited. These mutations are not limited to the heart mitochondria and may also be demonstrated throughout the liver, kidney, pancreas, central nervous system, skeletal muscles, and thyroid gland. The mitochondria are typically irregular, vary in size, and have a very abnormal cristae structure (Fig. 89.8). The myocardial changes can mimic HCM. Mitochondrial cardiomyopathy may also present with signs, symptoms, and features of DCM. Progressive hypertrophy and dilation of the heart with cardiac dysrhythmias are characteristics of these mitochondrial abnormalities. DNA defects frequently result in cytochrome c oxidase deficiency and abnormalities in the mitochondrial respiratory chain. Research is active in identifying these mutations by endomyocardial biopsy (45). Several systemic diseases are associated with mitochondrial dysfunction. For example, the MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) is complicated by cardiomyopathy and generalized microangiopathic occlusive disease (46). The wide spectrum of mitochondrial diseases is not limited to the heart; in fact, they are commonly accompanied by manifestations in other organ systems (47,48).
Specific Cardiomyopathies
Ischemic
Ischemic cardiomyopathy, defined as an ejection fraction of less than 40%, with multifocal wall motion abnormalities owing to coronary artery diseases, is the most common DCM in the United States (Fig. 89.9). It carries with it significant disability and mortality. In the past, a 5-year survival rate of only approximately 40% has been reported (49). Adequate perfusion of the myocardium is essential for appropriate cellular aerobic respiration, and heart failure due to ischemic cardiomyopathy is caused by at least four distinct mechanisms. Stunned and hibernating myocardial tissue are viable, with potential for improvement in functioning with better perfusion. Differentiation of ischemia, scar, and hibernating myocardial tissue is discussed in Chapters 50 and 55, and includes methods such as positron emission tomography, dobutamine and myocardial contrast echocardiography, and rest-redistribution thallium-201 scintigraphy (50). Diffuse small-vessel atherosclerosis can also produce ischemia-related malfunction of the cardiac
myocyte. Indeed, small vessel disease is involved in the pathogenesis of the cardiomyopathy of diabetes mellitus. Thus, diabetic cardiomyopathy may be due, in part, to epicardial vessel disease as well as diffuse small-vessel subendocardial atherosclerosis. Systemic lupus erythematosus and other autoimmune chronic inflammatory disease states occasionally produce similar findings.
myocyte. Indeed, small vessel disease is involved in the pathogenesis of the cardiomyopathy of diabetes mellitus. Thus, diabetic cardiomyopathy may be due, in part, to epicardial vessel disease as well as diffuse small-vessel subendocardial atherosclerosis. Systemic lupus erythematosus and other autoimmune chronic inflammatory disease states occasionally produce similar findings.
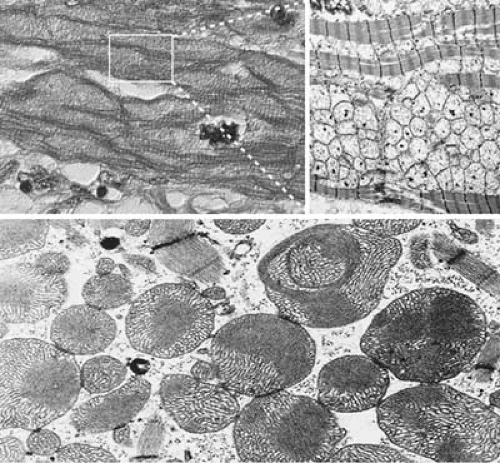 FIGURE 89.8. Mitochondrial cardiomyopathy. The top left image is a light micrograph of a cardiac myocyte in which the myofibrils (striations) are separated by granular material present throughout the sarcoplasm. The top right image is an electron micrograph from an area like the one shown in the white frame. Mitochondria are markedly increased in number and spreading apart the contractile myofibrils. The lower panel shows another case in which the mitochondria are pleomorphic in size and shape with very abnormal cristae. Some of them have “fingerprint” patterns and others show darker, electron-dense areas with paracrystalline inclusions. |
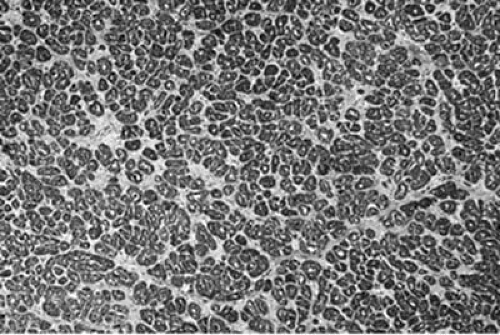 FIGURE 89.9. Chronic ischemic heart disease. This micrograph shows a cross-section of a bundle of cardiac myocytes and an increase of extracellular matrix, representing interstitial fibrosis, which surrounds and separates individual myocytes. |
 FIGURE 89.10. Marked concentric hypertrophy of the left ventricle. Notice the virtual absence of LV cavity owing to the massive hypertrophy of the wall and papillary muscles. |
Differentiation of ischemic cardiomyopathy from nonischemic processes is extraordinarily important. Patients with ischemic cardiomyopathy frequently, but not always, have histories of chest discomfort, myocardial infarctions, or abnormal ECGs, suggesting previous infarction. Restoring adequate perfusion to the heart is key to reversing the progression of a clinical heart failure state. Coronary angiography is key, although often lesions are seen that are not producing ischemia, and, on the other hand, diffuse small-vessel disease may not be well appreciated. Relating angiographic findings to perfusion, viability, and functional studies of the heart differentiates contributions of ischemia, scar, hibernation, and periinfarction stunning to the pathologic remodeling process of heart failure.
Valvular
Valvular cardiomyopathy is produced by valvular defects of numerous etiologies, discussed in Chapters 22,23,24. Typically, defects that produce volume-overloaded states (regurgitation) are more likely to cause cardiomyopathy than are lesions associated with pressure overload (valvular stenosis). Frequently, the issue of corrective valvular surgery in the setting of severe myocardial dysfunction is raised, with the hope of promoting myocardial recovery if the lesion is fixed. For instance, attempts at reducing mitral regurgitation have been performed in patients with severe cardiomyopathy, either as an isolated surgery or in conjunction with partial left ventriculectomy, with some symptomatic success and at least early improvement in certain cases (51,52).
Hypertensive
Hypertensive cardiomyopathy is discussed in detail in Chapter 7. Occasionally, patients with hypertensive cardiomyopathy can present with heart failure and a dilated heart. However, most patients show marked symmetric hypertrophy of the left ventricle (Fig. 89.10).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


