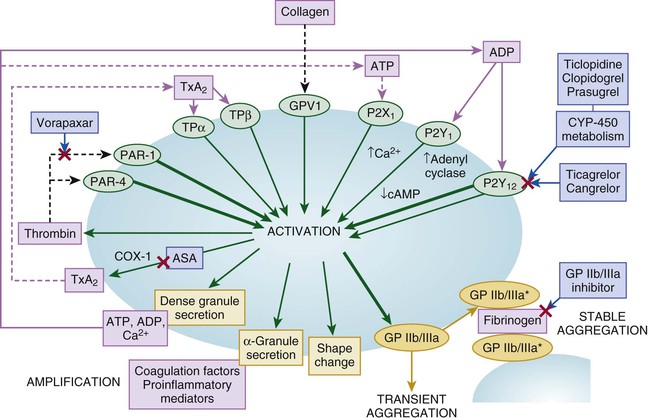
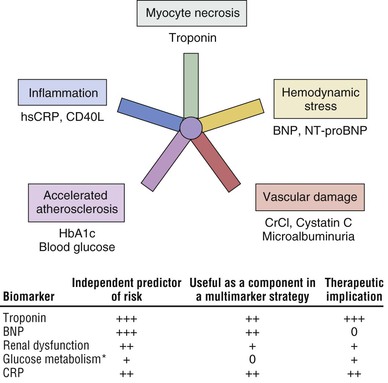
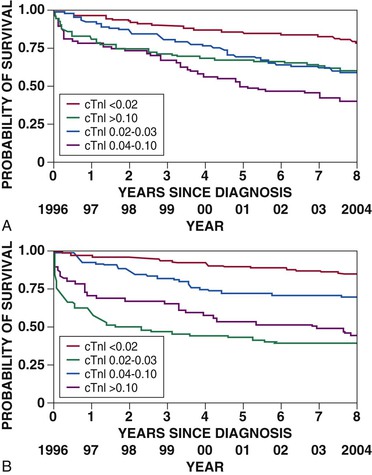
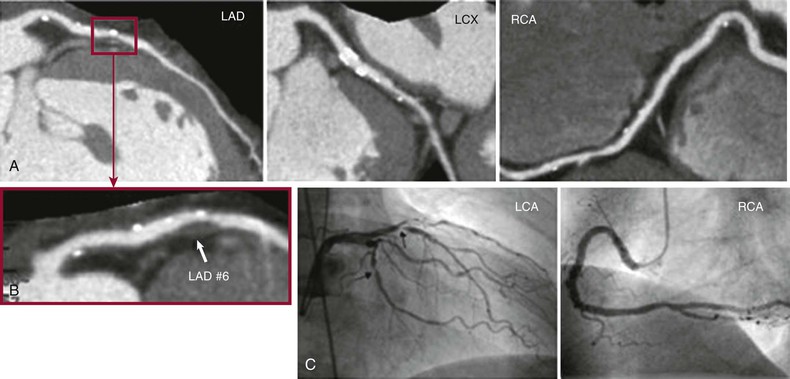
Robert P. Giugliano, Christopher P. Cannon, Eugene Braunwald Ischemic heart disease may be manifested clinically as either chronic stable angina (see Chapter 54) or an acute coronary syndrome (ACS). The latter, in turn, can be subdivided into ST-segment elevation myocardial infarction (STEMI), non–ST-segment elevation myocardial infarction (NSTEMI), or unstable angina (UA) (Fig. 53-1). Chapters 51 and 52 discuss STEMI in detail. Because NSTEMI and UA are indistinguishable at initial evaluation and the entity of UA is receding as the sensitivity of biomarkers of myocardial injury increases, they are often described together as NSTE-ACS and are discussed together in this chapter. Features that help differentiate ACS from stable angina are (1) onset of symptoms at rest (or with minimal exertion) and lasting longer than 10 minutes unless treated promptly; (2) severe, oppressive pressure or chest discomfort; and (3) an accelerating pattern of symptoms that develop more frequently, occur with greater severity, or awaken the patient from sleep. Symptoms alone do not suffice to distinguish the three types of ACS from one another. Patients without persistent (>20 minutes) ST-segment elevation in two or more contiguous leads but with biomarker evidence of myocardial necrosis are classified as having NSTEMI, whereas in patients without such evidence of myocardial necrosis, UA is diagnosed—a condition generally carrying a better prognosis. Globally, ischemic heart disease remains the number one cause of mortality; it was responsible for 7 million of the 53 million deaths reported in 2010.1 ACS, the acute manifestation of ischemic heart disease, accounted for approximately 1.1 million discharges in the United States in 2009,2 with approximately twice this number in Europe. The annual number of hospital discharges for ACS in developed countries has declined slowly over the past two decades, accompanied by an increase in nations with developing economies (see Chapter 1).3 In the United States, three recent trends have changed the frequency distribution of the types of ACS: (1) wider use of primary preventive therapies (aspirin, statins, smoking cessation) appears to have resulted in fewer cases of STEMI4; (2) aging of the U.S. population, with higher rates of diabetes and chronic kidney disease (CKD), have increased the incidence of NSTE-ACS5; and (3) the use of more sensitive assays for myocardial necrosis (i.e., cardiac-specific troponin [cTn]) has shifted the classification of NSTE-ACS away from UA toward NSTEMI5 (Fig. 53-2; also see Fig. 51-2A). Overall, the age- and sex-adjusted incidence rates of NSTEMI have grown slowly since 1999.4 The pathogenesis of NSTE-ACS involves four processes: (1) rupture of unstable atheromatous plaque, (2) coronary arterial vasoconstriction, (3) imbalance between the supply and demand of the myocardium for oxygen, and (4) gradual intraluminal narrowing of an epicardial coronary artery because of progressive atherosclerosis or poststent restenosis. These processes are not mutually exclusive and can occur simultaneously in any combination. Plaque rupture or erosion leads to the formation of superimposed thrombus (typically nonocclusive in NSTE-ACS) along with subsequent impaired myocardial perfusion, which if persistent, leads to myocardial necrosis. Inflammation of the arterial wall and the action of metalloproteinases produced by inflammatory cells in degrading the fibrous wall of plaque contribute to their instability (see Chapter 41). Vasoconstriction causing dynamic obstruction of coronary arterial flow may result from spasm of the epicardial coronary arteries (Prinzmetal angina, see below)—constriction of small, intramural, muscular coronary arteries resulting in increased coronary vascular resistance. This constriction may result from vasoconstrictors released by platelets, endothelial dysfunction (cardiac syndrome X; see Chapter 77), or adrenergic stimuli (e.g., the “fight-or-flight” response, cold, cocaine, or amphetamines [Chapter 68]). More than one of these mechanisms may be present simultaneously. Insufficient myocardial O2 supply may also occur in patients with severe anemia and hypotension. When an increase in myocardial O2 demand (e.g., tachycardia, fever, thyrotoxicosis) occurs in a patient with fixed narrowing of an epicardial coronary artery, secondary NSTE-ACS may develop. Activation of the coagulation cascade and platelets plays a central role (described in detail in Chapter 82) in the formation of thrombus following plaque rupture/erosion. The first step in thrombus formation is vascular injury or endothelial dysfunction, which causes adhesion of platelets to the arterial wall via binding of platelet glycoprotein (GP) Ib to subendothelial von Willebrand factor. Exposure of platelets to subendothelial collagen and/or circulating thrombin causes platelet activation (Fig. 53-3), which induces platelets to change shape and results in degranulation with release of adenosine diphosphate (ADP) and thromboxane A2 (TxA2)—which in turn causes further platelet activation and expression of platelet glycoprotein GP IIb/IIIa. In parallel, tissue factor expressed within the lipid-rich core of atherosclerotic plaque, when exposed to circulating blood, activates the coagulation cascade. A complex of tissue factor and coagulation factors VIIa and Va leads to the formation of activated factor X (factor Xa), which in turn amplifies the production of activated factor IIa (thrombin). The cascade proceeds with thrombin-induced conversion of fibrinogen to fibrin. The platelet and coagulation systems converge in that thrombin is also a potent platelet activator. Platelet GP IIb/IIIa binds circulating fibrinogen, thereby causing platelet aggregation and ultimately producing a platelet-fibrin thrombus, portions of which may embolize distally and cause myocardial necrosis. The central role of coronary artery thrombosis in the pathogenesis of NSTE-ACS is supported by (1) autopsy findings of thrombi in the coronary arteries typically localized to a ruptured or eroded atherosclerotic plaque; (2) a high incidence of thrombotic lesions in coronary atherectomy specimens in patients with NSTE-ACS in comparison to those with stable angina; (3) observations of plaque ulceration and/or irregularities in the fibrous cap of atherosclerotic plaque consistent with plaque rupture and thrombus formation as visualized by coronary angiography, intravascular ultrasound (IVUS), optimal coherence tomography (OCT), or computed tomographic angiography (CTA); (4) elevation of serum markers of platelet activity, thrombin generation, and fibrin formation; and (5) improvement in clinical outcome with antiplatelet and anticoagulant treatment. NSTE-ACS resulting from atherosclerosis is relatively uncommon in men younger than 40 years and women younger than 50 years, but the incidence rises steadily thereafter. Although NSTE-ACS may be the initial manifestation of coronary heart disease (CHD), most patients have preceding stable angina or myocardial infarction (MI). Patients with ACS more frequently have traditional risk factors for CHD (see Chapter 42) than do normal subjects or those with nonischemic chest pain. Although coronary risk factors can be used to assess risk in populations, they are less helpful in the assessment of individual patients. The initial symptom is typically described as pressure, heaviness, or frank pain beneath the sternum (see Chapter 50), and it resembles stable exertional angina—but is usually more intense and lasts longer (>20 minutes). Associated radiation to the ulnar aspect of the proximal part of the left arm, either shoulder, the neck, or the jaw may occur, but symptoms may be present anywhere between the ear and epigastrum.6 Symptoms such as diaphoresis, nausea, abdominal pain, dyspnea, and syncope may accompany the pain. Features that support the diagnosis include exacerbation of symptoms by physical exertion; precipitation by severe anemia, infection, inflammation, fever, or metabolic or endocrinologic (e.g., thyroid) disorders; and importantly, relief with rest or nitroglycerin. Atypical manifestations, such as dyspnea without chest discomfort, pain limited to the epigastrium, or indigestion, represent “anginal equivalents.” These atypical findings are more prevalent in women, older adults, and patients with diabetes, CKD, or dementia and can lead to underrecognition, undertreatment, and worse outcomes. Chest pain that is pleuritic or described as stabbing is generally noncardiac in origin. The clinical manifestations may be sudden, with severe, new-onset symptoms occurring during minimal exertion (Canadian Cardiovascular Society class7 [CCSC] III) or at rest (CCSC IV), an accelerating pattern of angina (more frequent, more intense, longer lasting), or angina occurring shortly after a completed MI.8 Findings on physical examination may be normal, although patients with large territories of myocardial ischemia may have audible third and/or fourth heart sounds. Rarely, hypotension, pale cool skin, sinus tachycardia, or frank cardiogenic shock can occur; these findings are far more common with STEMI than with NSTE-ACS. The examination can also be important in that potential precipitating causes of ACS can be identified, such as fever, resistant hypertension, tachycardia, profound bradycardia, thyroid disease, or gastrointestinal bleeding. Finally, findings on physical examination such as pulse deficits, tachypnea, and tachycardia in the presence of clear lung fields and pulsus paradoxus with jugular venous distention may lead to alternative life-threatening diagnoses such as aortic dissection, pulmonary embolism, or cardiac tamponade. The most common abnormalities on the 12-lead electrocardiogram (ECG) are ST-segment depression and T wave inversion; they are more likely to be present while the patient is symptomatic. Comparison with a recent ECG is important because dynamic ST-segment depressions as little as 0.05 mV are a sensitive (albeit not very specific) marker for NSTE-ACS. Greater degrees of ST-segment depression predict poorer outcomes, however, even when adjusted for other prognostic factors.9,10 Transient ST-segment elevation lasting less than 20 minutes occurs in up to 10% of patients and suggests either coronary vasospasm or an aborted infarction. Deep (>0.2 mV) T wave inversions are compatible with, but not necessarily diagnostic of NSTE-ACS, whereas isolated T wave inversions of lesser magnitude are not particularly helpful given their low specificity. In patients with definite NSTE-ACS, findings on the ECG may be normal or nondiagnostic in more than half of patients. Because ischemia may occur in a territory that is not well represented on the standard 12-lead ECG (see below) or because the patient may have episodic ischemia that is missed on the initial ECG, tracings should be repeated every 20 to 30 minutes until the symptoms resolve, the diagnosis of MI is established or excluded, or an alternative diagnosis is made. Coronary angiography identifies a culprit lesion in the circumflex coronary artery in a third of patients with high-risk NSTE-ACS.11 Because the standard 12-lead ECG does not represent this territory well, assessment of posterior leads V7 through V9 should be considered in patients with a history suggestive of ACS and a nondiagnostic initial ECG. Similarly, ACS caused by isolated involvement of an acute marginal branch of the right coronary artery is often not apparent on the standard 12-lead ECG but may be suspected from leads V3R and V4R.12 Therefore it is useful to obtain these extra leads in patients suspected of having ACS but with normal findings on a 12-lead ECG. Continuous monitoring of the ECG in the days following NSTE-ACS can identify patients at higher risk for recurrent events. ST-segment depressions noted on such monitoring within the first week after NSTE-ACS are associated with an increased risk for reinfarction and death.13 Biomarkers reflecting the pathogenesis of NSTE-ACS aid in diagnosis and prognosis. They include markers of myocyte necrosis, hemodynamic perturbation, vascular damage, accelerated atherosclerosis, and inflammation (Fig. 53-4). During the past decade, cardiac-specific troponins (cTnI and cTnT) have become the biomarkers of choice to identify myocardial necrosis and hence distinguish NSTEMI from UA. Several pathobiologic mechanisms can lead to the release of detectable levels of cTn in blood (Table 53-1). Because of differences among assays, there is consensus that the diagnosis of acute MI requires an elevation in cTnI or cTnT above the 99th percentile of the normal range for the specific assay used,14 a typical temporal rise and decline when serial samples are tested, and a clinical picture consistent with ACS. TABLE 53-1 Mechanisms of Troponin Release * Diseased skeletal muscle may also cause increases in circulating cTnT (but not in cTnI). From White HD: Pathobiology of troponin elevations: Do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol 57:2406, 2011. Although elevated cTn usually reflects myocardial necrosis, it does not always reflect MI. Abnormal elevations have been observed with a number of conditions, including heart failure, pulmonary embolism, myocarditis, pericarditis, transplant rejection, chemotherapy, and direct or indirect cardiac trauma. Furthermore, cTnI may be elevated chronically at a low-grade level in patients with severe CKD (stages IV and V), and all cTn is cleared more slowly in patients with impaired renal function. Therefore interpretation of the clinical significance of elevated cTn in such patients requires care. In fact, an estimated 60% to 70% of individuals with chest discomfort seen in an emergency department will have measurable cTn concentrations,15 but only a minority of them are experiencing acute MI. Patients with clinical findings suggestive of NSTE-ACS should have serial measurements of cTn beginning at initial evaluation. Newer high-sensitivity assays available in Europe (but not in the United States as of 2012) can exclude myocardial necrosis if two values measured 3 hours apart are both normal.16 However, the specificity of high-sensitivity cTn assays in the diagnosis of NSTE-ACS may be as low as 60%, even in patients with established CHD.17 The fourth-generation cTn assays currently used in the United States are less sensitive than the so-called high-sensitivity assays, and two negative cTn assays at least 6 to 9 hours apart are needed to exclude MI. The change in cTn between measurements appears to be more important than the actual concentration and can help distinguish MI from other processes that cause elevated cTn.18 In addition, normal early cTn levels are useful in the identification of patients at very low risk for cardiovascular events over the next 6 months19; they are useful in long-term prognostication as well (Fig. 53-5). As already noted, an important consequence of the use of increasingly sensitive cTn assays is that the fraction of NSTE-ACS patients with UA has decreased in favor of NSTE-MI. This reclassification is important because even minor elevations in cTn are associated with poorer outcomes than in patients without evidence of myonecrosis. Reclassification from UA to NSTEMI can lead to more aggressive treatment of patients with a low-level, “positive” cTn assay.20 Other biomarkers also increase in the days to weeks following NSTE-ACS. Natriuretic peptides (i.e., brain natriuretic peptide [BNP] and N-terminal pro-BNP) rise in proportion to the degree of ventricular distention and correlate with the risk for adverse events.21,22 In patients with NSTE-ACS, a baseline BNP measured on average 40 hours after the onset of symptoms correlated strongly with risk for death, heart failure, and MI through 10 months in a graded fashion.21 Baseline natriuretic peptide levels also help identify patients more likely to benefit from more aggressive treatments, including intensive anti-ischemic regimens,22 aggressive statin therapy,23 and early coronary revascularization.24 C-reactive protein (CRP) is a marker of inflammation that is elevated following ACS, and persistently elevated levels after discharge are associated with increased long-term cardiovascular risk. Elevated levels of fasting blood glucose and glycosylated hemoglobin indicate the presence of diabetes mellitus or metabolic syndrome and portend accelerated atherosclerosis and an increased risk for cardiovascular events in both the short and long term.25 Renal dysfunction, as reflected by elevated levels of cystatin C and creatinine, is associated with an increase in cardiovascular events, including cardiovascular mortality, in patients with NSTE-ACS. Several novel biomarkers can help improve prognostication in patients with NSTE-ACS (Table 53-2). These biomarkers tend to fall into two general categories: (1) markers that predict death and/or ischemic events26–33 and (2) markers that predict heart failure.34–36 TABLE 53-2 Emerging Biomarkers in Acute Coronary Syndromes Multimarker approaches involving biomarkers that are independent predictors of outcome are increasingly being used.37 One such approach uses three common biomarkers (cTn, CRP, and BNP), with an increasing number of abnormal markers correlating with a stepwise higher risk for subsequent ischemic complications.38 Measurement of serum lipids, including low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol and triglyceride, is useful in identifying important, treatable risk factors for coronary atherothrombosis (see Chapter 45). The first available sample should be used to guide therapy. Evaluation for other secondary causes of NSTE-ACS39 may also be appropriate in selected patients (e.g., determining the presence of hypoxemia, anemia, and disturbed thyroid function) because such “secondary” NSTE-ACS can often be treated and recurrences prevented. Goals of noninvasive testing in patients with suspected NSTE-ACS include (1) determining the presence or absence of coronary artery disease (CAD); (2) establishing CAD as the cause of the elevated cTn in patients with other possible explanations; (3) evaluating the extent of residual ischemia after medical therapy has been initiated, thus guiding further therapy; (4) localizing the ischemia before a planned percutaneous coronary intervention (PCI) in patients with multivessel disease; and (5) assessment of left ventricular function. The safety of early stress testing in patients with NSTE-ACS has been debated, but pharmacologic or symptom-limited stress testing appears to be safe after a period of at least 24 hours of stabilization without symptoms of active ischemia40; contraindications include active ischemia or other signs of hemodynamic or electrical instability. The merits of various modalities of stress testing have been compared (see Chapter 13). Exercise stress myocardial perfusion imaging with sestamibi (Chapter 16) and stress echocardiography with dobutamine have slightly more sensitivity than electrocardiographic exercise stress testing does alone. A useful approach is to individualize the choice based on patient characteristics, local availability, and expertise in interpretation. For most patients, electrocardiographic exercise stress testing is recommended if the ECG at rest lacks ST-segment abnormalities. If ST abnormalities exist at rest or if the patient is unable to exercise or cannot achieve a significant workload during exercise, pharmacologic stress perfusion or echocardiographic imaging is recommended. Findings consistent with high risk (e.g., severe ischemia as reflected by ST-segment depression ≥0.2 mV, hypotension, ventricular tachyarrhythmia, new or worsening left ventricular dysfunction) are indications to proceed rapidly with coronary angiography with the intent of performing revascularization if the coronary anatomy is appropriate. Echocardiography is useful in the assessment of left ventricular systolic and diastolic function and can also be used to identify left atrial dilation,41 functional mitral regurgitation,42 tricuspid annular plane systolic excursion,43 diastolic dysfunction,44 ventricular mechanical dyssynchrony,45 and ultrasound lung comets43 (extravascular lung fluid observed on thoracic ultrasound scanning)—each of which has been associated with an adverse prognosis in patients with NSTE-ACS. Contrast-enhanced coronary CTA (CCTA) in patients with or suspected of having NSTE-ACS can help establish the diagnosis of epicardial CAD in patients with equivocal signs and symptoms and identify unstable plaque at high risk for rupture. Detailed analysis of plaque morphology has identified two characteristics—positive vessel remodeling and low-attenuation plaque of lipid-rich lesions (Fig. 53-6)—that were associated with plaque rupture and ACS in 1059 patients observed for clinical events for an average of 27 months after imaging.46
Non–ST Elevation Acute Coronary Syndromes
Background
Definitions
Epidemiology
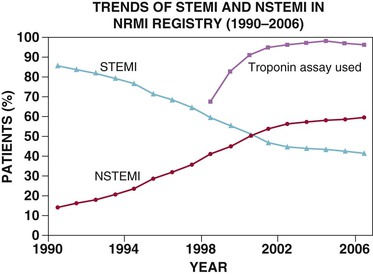
Pathophysiology
Clinical Assessment
History and Physical Examination
Physical Examination
Electrocardiography
Laboratory
TYPE
EXAMPLES/EXPLANATION
Myocyte* necrosis
Ischemia, infarction, inflammation, infiltration, trauma, toxic/metabolic (e.g., sepsis)
Apoptosis
Programmed cell death because of activation of caspases
Normal myocyte turnover
Natural low-grade annual turnover of myocytes (unclear whether this can be detected in the systemic circulation with current assays)
Cellular release of proteolytic troponin degradation products
Creation of small fragments that pass through the intact myocyte membrane without cell death
Increased cellular wall permeability
Reversible injury to myocyte membranes resulting in altered permeability (e.g., secondary to stretch, ischemia)
Formation and release of membranous blebs
Active secretion of vesicles or membrane expression with shedding (e.g., secondary to hypoxia)
MARKER NAME
DESCRIPTION
REFERENCE
Markers That Predict Death and/or Ischemic Events
Growth differentiation factor-15
Member of the transforming growth factor-beta cytokine superfamily that is released from cardiomyocytes after ischemia and reperfusion injury
26
Heart-type fatty acid–binding protein
Cytoplasmic protein involved in intracellular uptake and buffering of free fatty acids in the myocardium
27
Myeloperoxidase
A hemeprotein released during degranulation of neutrophils and some monocytes
28
Pregnancy-associated plasma protein A
Zinc-dependent matrix metalloproteinase abundantly expressed in eroded and ruptured plaque but only minimally expressed in stable plaque
29
Placental growth factor
Member of the vascular endothelial growth factor family that is strongly upregulated in atherosclerotic lesions and acts as a primary inflammatory instigator of atherosclerotic plaque instability
30
Secretory phospholipase A2
Hydrolyzes phospholipids to generate lysophospholipids and fatty acids, thereby enhancing susceptibility of the vessel to atherogenesis
31
Interleukin-6
Stimulator of hepatic synthesis of C-reactive protein
32
Chemokine ligand-5 and ligand-18
Mediators of monocyte recruitment induced by ischemia
33
Markers That Predict Heart Failure
Midregional proadrenomedullin
Peptide fragment of the vasodilatory peptide adrenomedullin
34
Neopterin
Marker of monocyte activation
35
Osteoprotegerin
Modulator of immune function and inflammation
36
Noninvasive Testing
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Non–ST Elevation Acute Coronary Syndromes
53
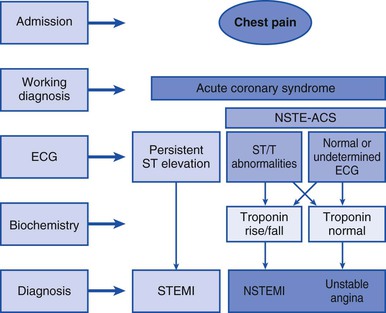
FIGURE 53-2 Trends of STEMI and NSTEMI in National Registry of Myocardial Infarction (NRMI) from 1990 to 2006. The proportion of patients with STEMI or NSTEMI and the proportion of patients in whom a troponin assay was used to diagnose AMI are shown. (From Rogers WJ, Frederick PD, Stoehr E, et al: Trends in presenting characteristics and hospital mortality among patients with ST elevation and non–ST elevation myocardial infarction in the National Registry of Myocardial Infarction from 1990 to 2006. Am Heart J 156:1026, 2008.) (Also see Figure 51-2.)