Fig. 23.1
Chest radiograph from a patient with acute interstitial pneumonia demonstrating diffuse bilateral infiltrates without focal consolidation
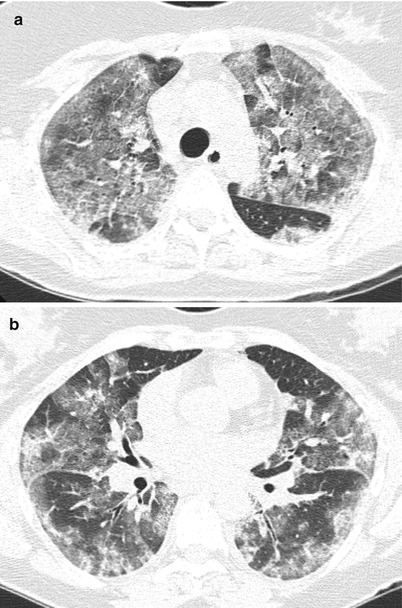
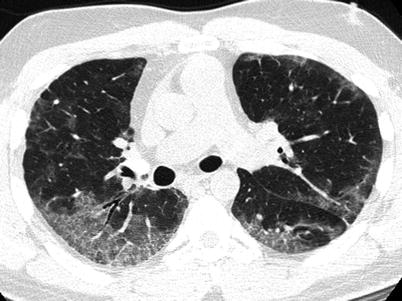
Fig. 23.2
High resolution computed tomography images from the upper (panel a) and mid (panel b) lung fields demonstrating diffuse bilateral ground glass opacifications without consolidation or effusions
Her labs were notable for a leukocytosis with a white blood cell count of 14,000 and differential showed 82 % neutrophils. Urinalysis showed no evidence of infection, and blood cultures were negative. A transthoracic echocardiogram showed no evidence of increased pulmonary arterial or right ventricular pressure, and no left ventricular dysfunction. On her fourth day of hospitalization, given her lack of improvement on broad-spectrum antibiotics, she was started empirically on one gram of methylprednisolone per day. A lung biopsy was performed on her sixth day of hospitalization, and revealed diffuse alveolar damage.
History and Definition
Acute interstitial pneumonia (AIP) – also called Hamman-Rich syndrome – is a rapidly progressive diffuse parenchymal lung disease of unknown etiology resulting in hypoxemic respiratory failure which is often fatal. The presentation – both clinically and radiographically – is virtually identical to Acute Respiratory Distress Syndrome (ARDS) but occurs in the absence of shock or infection [1].
By definition, AIP is idiopathic and thus granulomatous diseases, autoimmune diseases, infections or other exposures that could cause interstitial lung disease (ILD) must be excluded.
Epidemiology
Acute interstitial pneumonia is a relatively rare but probably underdiagnosed disease. Review of the literature suggests that an individual academic institution may diagnose only a few cases a year, if any [2, 3]. AIP is often a diagnosis of exclusion and requires biopsy confirmation; therefore it is possible that there is a subset of self-limited cases that improve with supportive care and go unrecognized. Conversely, there may be fatal cases that are misclassified as ARDS or pneumonia that never undergo confirmatory biopsy [3].
Age of onset is quite variable, and both pediatric patients as well as the elderly can be affected [4]. No clear risk factors have been convincingly identified in the literature – specifically there is no known link with tobacco exposure – and many of those affected are in good health with no comorbidities.
Presentation
Presenting symptoms vary from patient to patient, but often include cough, dyspnea and occasionally fever. Many patients present with symptoms mistaken for a viral upper respiratory tract infection [5, 6].
Shortness of breath is a hallmark feature and is eventually present in virtually all cases. Given the relatively non-specific historical features, this condition is commonly misdiagnosed early on as pulmonary embolism, infection or congestive heart failure [2].
Physical exam findings are relatively non-specific, including diffuse crackles. Virtually all patients with fulminant AIP are too ill to perform pulmonary function testing, but they would likely reflect diffuse alveolar and bronchiolar damage, showing a restrictive pattern with reduced DLCO [7]. All patients should be examined for joint deformities or skin changes that might suggest the presence of connective tissue disease.
Diagnostic Evaluation
Thorough evaluation for infectious etiologies – particularly viral infections and atypical infections such as pneumocystis pneumonia, legionella, mycoplasma and fungal infections – should be pursued. Cultures of blood and sputum, as well as bronchoalveolar lavage (BAL), are frequently performed. A transthoracic echo to evaluate for left ventricular dysfunction is often obtained. Most patients have a leukocytosis at time of presentation [6].
Bronchoalveolar lavage (BAL) findings are indistinguishable from ARDS and typically include increased total cells, red blood cells, hemosiderin and neutrophils [8]. These findings while supportive are not diagnostic, and the chief utility of a BAL is to exclude other etiologies of respiratory failure and exclude infection. Bronchoalveolar lavage (BAL) can also exclude pulmonary hemorrhage, especially in a patient where a systemic vasculitis or connective tissue disease is suspected.
A thorough medical history should be taken from the patient or family members to clarify whether the patient has received any chemotherapy, has had any previous radiation exposures or whether there is any past history of connective tissue disease. A positive rheumatoid factor in a patient with the appropriate phenotype might be worrisome connective tissue associated-ILD. A positive dsDNA or anti-smith antibody would be consistent with SLE, whereas a negative ANA is helpful in excluding SLE as an underlying etiology. Positive anti-Scl-70 would be concerning for systemic scleroderma. Positive anti-Jo antibodies in conjunction with elevated CPK, Aldolase and ALT would be consistent with an anti-synthetase syndrome which can manifest with rhabdomyositis and ILD. A social history to evaluate for any possible drug or occupational exposures is also warranted.
Radiology
Common findings on high resolution computed tomography (HRCT) in AIP are similar to ARDS, including ground-glass attenuation, air-space consolidation, interlobular septal thickening and traction bronchiectasis. Patients with AIP are more likely to have a distribution of disease that is largely symmetric with a predeliction for lower lung fields compared to ARDS [9]. Most patients have involvement that is quite diffuse, involving 45–95 % of lung parenchyma [3]. There is some evidence that the presence of traction bronchiectasis may reflect fibrosis and portend a worse outcome [10].
Histopathology
Establishing a histopathologic diagnosis is requisite for diagnosing AIP, and furthermore can be helpful in excluding other more treatable disease states such as sarcoid or infection. Open lung biopsy will best demonstrate the architectural and histopathologic changes. Given the diffuse nature of the disease, however, AIP is one of the few interstitial pneumonias in which transbronchial biopsy can be helpful as even small transbronchial specimens may be sufficient to capture the needed pathologic characteristics [1, 7]. Many diagnoses are made on autopsy [10].
Although helpful from an academic standpoint, there is very little in the way of therapeutic options available for AIP which means obtaining a tissue diagnosis may not provide much benefit depending on the clinical scenario. If infection has already been excluded with BAL, many practitioners would trial empiric steroids rather than pursuing a biopsy-confirmed diagnosis. One can imagine specific scenarios where the risks of a biopsy would be too high to the patient to warrant the procedure, whether secondary to clinical instability or bleeding risk. Alternatively, there may be instances in which a definitive diagnosis might alter goals of care. This highlights the importance of determining whether to pursue histopathologic diagnosis on a case-by-case basis.
The histopathologic hallmark of AIP is diffuse alveolar damage. Alveoli containing inflammatory cells – lymphocytes, plasma cells and macrophages with occasional neutrophils – are often seen. The remnants of hyaline membranes within alveoli are frequently observed. Other key features on biopsy are interstitial fibrosis and edema, as well as the presence of type II pneumocyte hyperplasia [5] (Fig. 23.3).
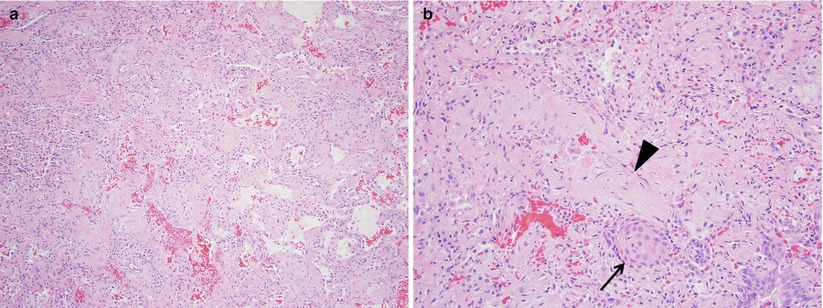
Fig. 23.3
At low power (panel a), acute interstitial pneumonia is characterized by diffuse interstitial thickening with associated alveolar septal collapse. At higher magnification (panel b), subepithelial proliferations of fibroblasts and myofibroblasts in a myxoid collagen background are common (arrowhead) and residual hyaline membranes are usually not prominent. Squamous metaplasia (arrow), although a nonspecific finding, is common in acute interstitial pneumonia
A key feature that distinguishes AIP from more chronic processes is the presence of extensive fibroblast proliferation and relative absence or paucity of collagen deposition. Another helpful differentiating characteristic is the relative uniformity of disease from field to field, as compared to a chronic interstitial pneumonia, which reflects multiple insults with varying degrees of inflammation and fibrosis [5]. Architectural distortion, when observed, is generally associated with a poor prognosis.
Clinical Course
Onset of symptoms prior to rapid deterioration is somewhat variable, but patients typically have vague symptoms for 1–2 weeks before progressing to the point of requiring admission to an intensive care unit [2]. Most patients become rapidly ventilator dependent secondary to refractory hypoxemia. The mortality rate is greater than 50 %, and average time to death averages approximately 2 weeks [2, 5, 7]. The minority of patients who survive AIP generally do well in the absence of other comorbidities. While there have been some reports of recurrent AIP and many patients go on to develop ILD, some patients do not have any clinically significant pulmonary sequellae [3, 6].
Pathophysiology
Similar to ARDS, AIP is characterized by diffuse alveolar damage comprised of an exudative phase, early proliferative phase and a late proliferative phase, which in some cases evolves to extensive fibrosis [11].
In the exudative phase, acute alveolar epithelial damage results in the release of inflammatory mediators and recruitment of macrophages, lymphocytes, plasma cells and neutrophils. This overwhelming immune response results in interstitial edema and further epithelial injury. The initial robust immune response is followed by an organizing phase characterized by fibroblast proliferation and thickening of the alveolar septae [6]. The rapidity of progression varies from patient to patient, but typically the pattern of injury is uniform, reflecting a simultaneous insult rather than multiple insults at different points in time.
Treatment
Initial treatment is both empiric and supportive. All patients should receive lung-protective ventilation, as this is appropriate for most diagnoses on the differential. Broad spectrum antimicrobial coverage is appropriate until infection is satisfyingly excluded, usually with BAL.
Once the diagnosis is established, however, there is unfortunately insufficient data for any evidence-based therapy. In addition to lung-protective ventilation strategies, many clinicians trial a course of corticosteroids [3, 7, 12]. Some experts have suggested methylprednisolone 1 g/day IV for 3 days followed by 1 mg/kg/day IV or oral prednisolone for 4 weeks with subsequent tapering. There is limited evidence that patients who receive steroid therapy during the acute, exudative phase of their disease fare better than those whose lung biopsies already demonstrated proliferation and architectural distortion [3].
Respiratory Bronchiolitis Interstitial Lung Disease (RB-ILD)/Desquamative Interstitial Pneumonia (DIP)
Clinical Vignette
A 38-year-old man presents to his primary care clinic complaining of a persistent dry cough for approximately 1 year. He complains of mild dyspnea on exertion that does not limit any of his activities. Additionally, he denies any fevers or chills and has not been exposed to any sick contacts. The patient is a current smoker of 1.5 packs per day and has been doing so for 21 years. On exam, his vital signs are normal and he is not hypoxic. He has diffuse bilateral dry crackles but the rest of his physical exam is entirely normal. His primary care physician orders a chest x-ray that shows upper lung predominant reticulonodular opacities. At this point the patient is referred to a pulmonologist who performs pulmonary function testing. A mild obstructive defect is seen with normal lung volumes and a decreased DLCO. A HRCT is performed and shows patchy GGO and centrilobular nodularity. Due to his persistent symptoms and abnormal imaging the patient undergoes a bronchoscopy with BAL and transbronchial biopsies. BAL cultures are unremarkable and cell count shows a normal differential. Biopsies reveal clusters of pigmented macrophages with associated fibrous scarring extending into the alveolar wall. A diagnosis of RB-ILD is made.
History and Definition
Respiratory bronchiolitis (RB) is a histopathologic lesion, seen in virtually all smokers, that was first recognized on autopsy series of young cigarette smokers who died of nonpulmonary causes. The histopathologic features are characterized by clusters of brown pigmented macrophages in the first-order and second- order respiratory bronchioles [13]. Clinically, RB is associated with asymptomatic or a minimally symptomatic disease state characterized by mild cough and/or dyspnea [13–15]. In rare cases, RB may present with significant pulmonary symptoms, abnormal imaging and pulmonary function testing. In this scenario it is described as RB associated interstitial lung disease (RB-ILD) [1].
Epidemiology
RB-ILD lies on a spectrum of smoking-related lung diseases that ranges from minimally symptomatic RB to very severe but pathologically similar desquamative intertstitial pneumonia (DIP). RB-ILD was first introduced by Myers et al as pathologic explanation for a clinically described chronic ILD where RB was the only pathologic diagnosis seen on biopsy [16, 17]. RB-ILD has since been further characterized as a distinct clinical entity, albeit very rare.
Presentation
The majority of patients present with nonspecific respiratory complaints including gradual onset of dyspnea and new or changing cough. Impairment in functional capacity is generally minimal and overlaps with early emphysema, making the diagnosis difficult. On exam, inspiratory crackles are the most prominent feature and are generally coarse in nature and occur throughout inspiration and occasionally into exhalation [15]. Rarely, do patients have any clubbing [1, 14, 15, 17]. RB-ILD is seen in patients in the fourth and fifth decades of life and the patients are usually heavy smokers (>30 pack years) with a 2:1 male predominance [1, 14, 17].
Diagnostic Evaluation
The pulmonary function testing (PFT) pattern most often seen in RB-ILD is obstructive, however, both restrictive and a mixed patterns are seen frequently as well. The fibrosis and inflammation of the respiratory bronchioles seen in RB-ILD are similar changes to those in COPD and along with coexisting emphysema are responsible for the obstruction [16]. However, a significant response to inhaled bronchodilators is not typically seen due to the fact that the pathologic process is not bronchospastic. Overall, the most common PFT abnormality is a decreased DLCO and often times will correlate with disease severity [1, 18]. Approximately, 10 % of patients have normal lung function at diagnosis [19].
There are no specific laboratory tests that aid in the diagnosis of RB-ILD. Disease severity can also correlate with imaging findings.
Radiology
Although there are no pathognomonic imaging findings associated with RB-ILD, common imaging patterns are seen. Chest radiography often appears normal but can have abnormalities such as thickening of the walls of the central or peripheral bronchi and fine reticulonodular opacities, typically diffuse or upper lung predominant [1, 14, 15]. HRCT is more clinically useful and frequently shows nonspecific changes such as centrilobular nodularity and patchy GGO with septal thickening (Fig. 23.4). The radiographic differential includes hypersensitivity pneumonitis and non-specific interstitial pneumonia (NSIP) [20]. Since all of these patients are smokers or former smokers, concomitant emphysema is common as well.
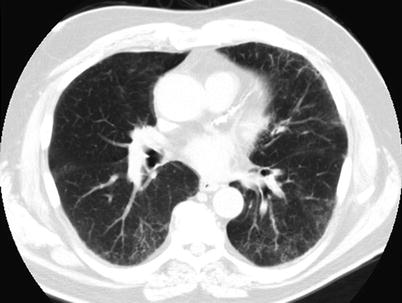
Fig. 23.4
High resolution computed tomography images from a patient with respiratory bronchiolitis interstitial lung disease demonstrating areas of faint, patchy ground glass opacification and reticular thickening
Histopathology
The next step in the work up of RB-ILD usually includes a bronchoscopy, as is the case with many ILDs. In the case of suspected RB-ILD bronchoscopy is not typically helpful. The vast majority of BAL samples are indistinguishable from lavage fluid from otherwise healthy smokers (increased cells with normal differential). One difference seen in RB-ILD is that there are a relatively higher amount of pigmented macrophages compared to healthy nonsmoking individuals [1, 18]. This finding is nonspecific and may be seen other smoking related lung diseases. A surgical lung biopsy is required to see definitive patterns of RB-ILD. Histopathologically RB-ILD differs from RB by demonstrating fibrous scarring that extends into the surrounding alveolar wall in addition to the aforementioned clusters of pigmented macrophages [16, 17]. These clusters are more frequently found near the bronchioles as compared to the rest of the lung parenchyma. Additionally, these clusters are more prominent than those seen in healthy cigarette smokers [15]. RB-ILD lacks features of usual interstitial pneumonia (UIP) such as honeycombing and fibroblastic foci. Concominant emphysema may be seen as well in RB and RB-ILD, owing to the strong association with smoking [16] (Fig. 23.5).
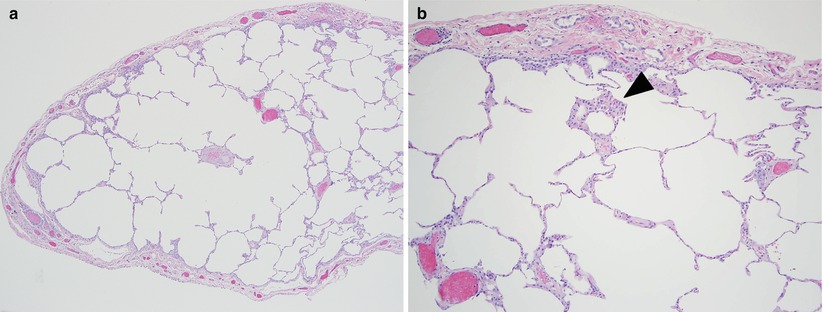
Fig. 23.5
The main histologic finding of respiratory bronchiolitis interstitial lung disease is similar to that of desquamative interstitial pneumonia; namely increased numbers of intraalveolar macrophages (panel a). This process is limited to the small bronchioles and peribronchiolar airspaces (arrowhead panel b) in respiratory bronchiolitis interstitial lung disease and is more diffuse in desquamative interstitial pneumonia, although no quantitative histologic criteria have been established. Histologically, desquamative interstitial pneumonia and respiratory bronchiolitis interstitial lung disease likely represent the ends of a continuum of smoking-related disease
Clinical Course
The natural history of RB-ILD is difficult to ascertain as it is very rare. Previous studies are conflicted as to whether patients worsen, improve or stabilize regardless of smoking status. Initial studies suggested it was a benign disease process however, more recent studies suggest that RB-ILD has a more sinister course [18, 21–23]. Additionally, there are reports of a disconnect between the patient’s described sense of stable or improved dyspnea and objective measurements of validated dyspnea scores which clearly worsened over the same time [18]. There have been no deaths reported due to RB-ILD.
Treatment
Treatment options for RB-ILD are limited but must include smoking cessation at the forefront. Studies are mixed regarding the reversibility of the disease process with smoking cessation with some studies arguing that there are significant improvement in symptoms, pulmonary function and imaging while others argue that there is merely stabilization [15, 18]. In some cases a trial of corticosteroids is used. Once again, results are mixed but recent studies suggest that there is no consistent improvement in symptoms or pulmonary function with corticosteroid treatment and therefore is not routinely recommended [18]. Empiric dosing of prednisone 0.5 mg/kg/day has been tried. The overall course is short (~4–6 weeks) and is usually reserved for patient with a documented decline in lung function despite abstinence from smoking [24]. It is unclear whether or not any intervention alters the natural history of the disease.
Desquamative Interstitial Pneumonia (DIP)
History and Definition
As mentioned above, DIP is another smoking related ILD thought to be on the severe end of the spectrum with RB-ILD. DIP was first described in 1965 as a case series of 18 patients noted to have similar pathology on open lung biopsy. This study defined a clinicopathologic syndrome characterized by a chest X-ray showing peripheral and basilar GGO and a clinical response to corticosteroid therapy with a combination of pathologic findings. These include extensive desquamation and proliferation of large alveolar cells within the alveolar space which contain PAS positive brown colored granules, accumulation of lymphoid follicles in the periphery of the lung, slight thickening of the alveoli without any evidence of necrosis, hyaline membranes or fibrinous exudates and uniform lesions throughout the lungs [25]. DIP was further expanded upon in 1978 by differentiating it from UIP [26]. In 1987, Myers et al. were the first to consider that RB-ILD may evolve into DIP [17]. The true relationship between DIP and RB-ILD is not well understood due to the rarity of both diseases. Since RB, RB-ILD and DIP are generally considered on the same spectrum of disease it is important to be able to differentiate between them. This distinction is especially important because each process has a different natural history. Specifically, there is increased morbidity and mortality associated with DIP as compared to RB and RB-ILD [27].
Epidemiology
Approximately 90 % of patients with DIP are smokers or former smokers although it can be seen with systemic disorders, infections and environmental triggers [22, 23, 26]. Due to its rarity and the inherent difficulties with disease recognition, it is difficult to make an accurate estimate of the incidence and prevalence of DIP. There are more than 150 reported cases in the literature [28]. The discovery of new cases of DIP has decreased recently. This is likely due to new classification systems from which cases that were previously described as DIP are now classified into other entities such as RB-ILD, NSIP or other diagnoses [24].
Presentation
Clinically, DIP behaves similarly to other ILDs, meaning, an insidious onset of dyspnea and cough over weeks to months. Presentation is usually in the fourth or fifth decades of life and has a male predominance, both similar to RB-ILD [1, 22]. On exam, most patients have inspiratory crackles and clubbing is common as well [1, 22, 23, 26]. The clinical distinctions between DIP and RB-ILD are subtle but notable as well with DIP causing generally more severe respiratory symptoms.
Diagnostic Evaluation
The work up for DIP, as is common with the other ILDs, includes a detailed history and physical exam, pulmonary function testing (PFT), chest radiography/HRCT and obtaining tissue specimens. On physical exam, clubbing is frequent in DIP but not seen in RB-ILD and pulmonary function testing can show obstruction in RB-ILD and this pattern is not seen in DIP [22, 23]. Pulmonary physiology may show normal lung volumes or varying amounts of restriction. This is consistent with the major pathology being a fibrotic process. The major and most consistent PFT abnormality is impairment in gas exchange signified by a low DLCO [23, 26].
Radiology
Chest radiography can be normal or show patchy abnormality including GGO or linear or reticulonodular infiltrates with a lower lung and peripheral predominance [1, 22, 26]. HRCT typically shows patchy GGO with a lower and peripheral lung zone predominance as well (Fig. 23.6). Irregular linear opacities are another common finding on HRCT. Honeycombing is uncommon but thin walled cystic changes can be seen within the areas of GGO [1, 14, 20, 22]. As these patients are current or former smokers, simultaneous emphysema may be present. In RB-ILD imaging studies show upper lung predominant disease whereas DIP is characteristically a lower lung and peripheral process. The HRCT differential includes RB-ILD, hypersensitivity pneumonitis, sarcoidosis, NSIP or atypical infection such as Pneumocystis jiroveci pneumonia [1, 14].