60 Neuromuscular Diseases and the Heart
Diseases of Muscle
Dystrophinopathies
Clinical manifestations of Duchenne’s MD become obvious at 3 to 5 years of age, with contractures and proximal muscle weakness greater than distal weakness. Gower’s maneuver is characteristic (Fig. 60-1). Nonprogressive mental retardation occurs in approximately 70% of patients. Severe scoliosis occurs in 90% by early in the second decade of life. There is steady progression to wheelchair use within 10 years, and death usually occurs in the second or early third decade of life from respiratory or cardiac failure. Cardiovascular manifestations include dilated cardiomyopathy, usually of the posterobasal and posterolateral left ventricle; mitral regurgitation; conduction abnormalities, most frequently incomplete right bundle branch block; atrial and ventricular arrhythmias; QT dispersion; and autonomic dysfunction manifested by abnormal heart rate variability.
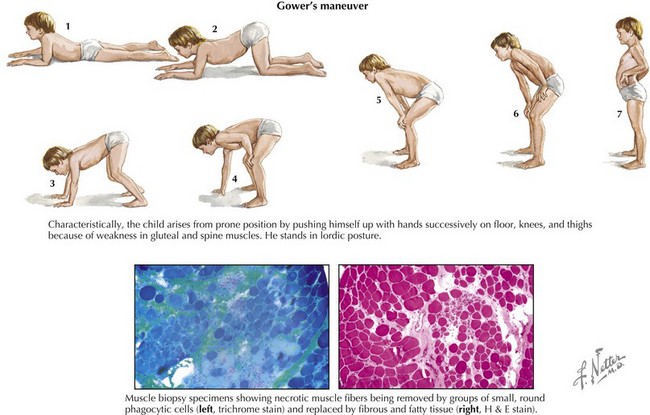
Both conditions result in marked, persistent CK elevation, generally 10 times the upper limit of normal or higher. The diagnosis can be made in more than 90% of patients by confirmation of a deletion or point mutation of the Xp21 gene locus. Analysis of dystrophin content by muscle biopsy is also a reliable diagnostic tool (see Fig. 60-1, lower).
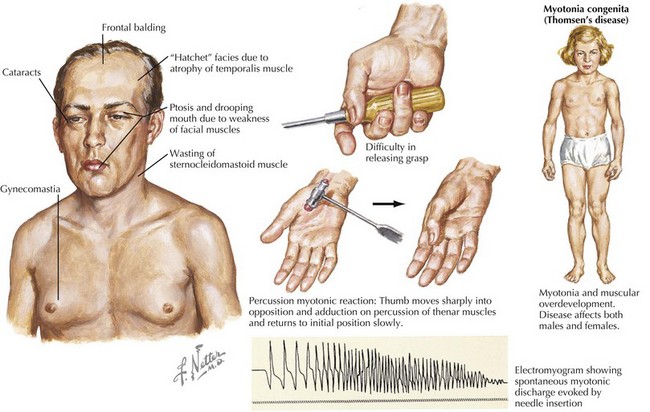
Myotonic Dystrophy
Myotonic dystrophy, an autosomal-dominant MD, is a multiple-system disease. Two types of myotonic dystrophy have been identified. Type 1, or DM1, is predominantly a distal myopathy caused by an expanding trinucleotide CTG-repeat of a specific protein kinase on chromosome 19. Type 2, or DM2, is a proximal myotonic dystrophy caused by the expansion of a CCTG repeat in the zinc finger protein (ZNF9) on chromosome 3. Disease expression varies within and between affected families. Patients may have facial muscle weakness, especially of the temporalis, levator palpebrae superioris, and masseters, giving typical “cadaveric” facies. Myotonia results in delayed muscle relaxation after contraction or muscle percussion (Fig. 60-2). Systemic features may include frontal balding, cataracts, hypogonadism, insulin resistance, dysphagia, hypersomnia, pickwickian syndrome, and mental retardation. Children of affected mothers with DM1 are more likely to be severely weak and hypotonic and to have mental retardation in infancy. Progression of myotonic dystrophy is variable, with death usually resulting from aspiration pneumonia, respiratory failure, or cardiac involvement.