NEONATAL CARDIOMYOPATHIES
Introduction
Cardiomyopathies are primary heart muscle disorders associated with systolic and/or diastolic cardiac dysfunction. A little over one-third of pediatric cardiomyopathies present with symptoms in the infantile period. Fetal and neonatal presentation of cardiomyopathies is relatively less common but carries a significantly worse prognosis with high mortality. An objective assessment of intrinsic abnormality of myocardial systolic function (contractility), diastolic function (relaxation and compliance), and growth (hypertrophy and atrophy) is important in classifying the cardiomyopathy into the phenotypic forms of dilated, hypertrophic, restrictive or mixed cardiomyopathy. A primary restrictive phenotype is rare in neonates.
Neonatal cardiomyopathies have significant etiologic heterogeneity with numerous genetic and acquired causes. The etiologies differ significantly compared to cardiomyopathies that present in older children, adolescents and adults. The diagnostic evaluation is complicated by a much wider spectrum of rare genetic causes, varied clinical presentations, and a complex array of specialized diagnostic tests and biochemical assays.
Cardiomyopathies with underlying genetic causes result in structural or metabolic derangements of cardiomyocytes. The involvement may be primary cardiac or may have additional multisystem abnormalities. Cardiomyopathies with acquired causes usually have infectious and/or immune mediated myocardial damage or in utero placental insult. Genetic malformation syndromes and metabolic disorders which result in abnormal storage of cellular metabolites and disorders of energy metabolism such as fatty acid oxidation and oxidative phosphorylation defects together account for almost one-third of cardiomyopathies that present in the neonatal/infantile period.
Phenotypic Classification of Neonatal Cardiomyopathies
The concept of “primary myocardial disorder” was first introduced in 1900, but it was not until 1957 that the term “cardiomyopathy” was first used. Since then the definition and classification of cardiomyopathies has continuously evolved, concurrent with an increasing recognition of the distinct morphologic phenotypes and underlying etiologic causes. The popular classification of cardiomyopathy includes 5 major morphologic subtypes: (1) Dilated Cardiomyopathy (DCM); (2) Hypertrophic Cardiomyopathy (HCM); (3) Restrictive Cardiomyopathy (RCM); (4) Left Ventricular Non-Compaction Cardiomyopathy (LVNC); and (5) Arrhythmogenic Cardiomyopathy (AC, also known as Arrhythmogenic Right Ventricular Cardiomyopathy, ARVC). (Figure 42.1).
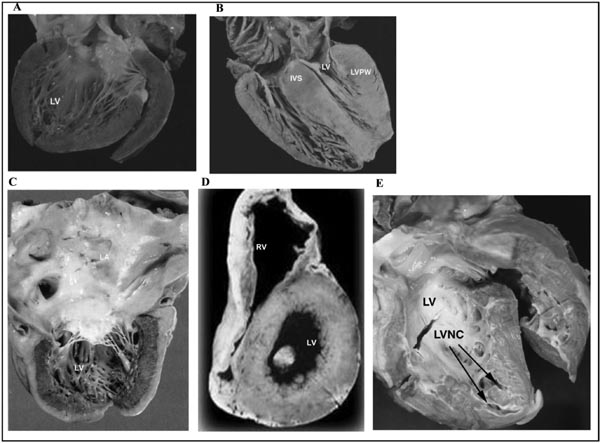
Figure 42.1. The five common phenotypic forms of cardiomyopathies: A: Dilated Cardiomyopathy (DCM), B: Hypertrophic Cardiomyopathy (HCM), C: Restrictive Cardiomyopathy (RCM), D: Arrhythmogenic Cardiomyopathy/ Arrhythmogenic Right Ventricular Cardiomyopathy (AC/ARVC), E: Left Ventricular Non Compaction (LVNC). Courtesy Dr. J.A Towbin, Cincinnati Children’s Hospital.
The most recent definition by the American Heart Association (AHA) in 2006 defines cardiomyopathies as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic”.1 In addition to the popular morphologic classification listed above, cardiomyopathies can be considered as “primary cardiomyopathies” which are solely or predominantly confined to heart muscle or “secondary cardiomyopathies” which show pathological myocardial involvement as part of a large number and variety of generalized systemic disorders.
The two most common phenotypes of neonatal cardiomyopathies are DCM and HCM,2 with an almost equivalent distribution between the two phenotypes or possibly a slight preponderance of HCM phenotype. Recently, LVNC has been recognized as a distinct form of cardiomyopathy and is being increasingly identified.3 RCM with primary diastolic dysfunction of both ventricles is rare in neonates. There is continued controversy whether endocardial fibroelastosis (EFE), a characteristic pathologic finding in several cases of pediatric and neonatal cardiomyopathies, is a distinct morphologic form of cardiomyopathy or just a pathologic finding in many cases of end-stage DCM.4
On several occasions, there may be a phenotypic overlap in the same patient giving a mixed cardiomyopathy phenotype as well as phenotype progression from one type to another over time. The same etiologic diagnostic group may be associated with multiple cardiomyopathy phenotypes. The descriptive classification of cardiomyopathy phenotypes as DCM or HCM does not denote the underlying etiology. Further characterization of the etiologic subtype is important in cause based therapy and prognosis.
Dilated Cardiomyopathy
DCM5 is characterized by dilatation and impaired contraction of the left ventricle (LV) or both ventricles6 and commonly results in congestive heart failure (CHF). The DCM heart appears globular in shape with significant biventricular dilation and usually has associated atrial enlargement with possible thrombi in the atrial appendages. The overall weight of the heart is increased due to compensatory myocardial hypertrophy. The histologic findings in DCM are non-specific and may include myocyte attenuation, myocyte hypertrophy, interstitial fibrosis, myocyte nuclear hypertrophy, and pleomorphism.7,8 DCM can be secondary to either genetic or acquired causes, with extensive genetic heterogeneity. The most common mode of inheritance is autosomal-dominant, followed by X-linked, matrilinear and autosomal-recessive inheritance patterns.
Endocardial fibroelastosis (EFE) is a distinct pathologic finding in some cases of DCM characterized by presence of glistening white fibroelastic tissue (consisting of collagen and elastic hyperplasia) over the endocardium with some subendocardial invasion, usually involving LV and left atrium (LA), and rarely, the right ventricle (RV).9
Hypertrophic Cardiomyopathy
HCM10 is characterized by left (LVH) and/or right ventricular hypertrophy (RVH), which may be asymmetric with predominant involvement of interventricular septum or symmetric concentric LVH, in the absence of an identifiable hemodynamic cause. There may or may not be associated left ventricular outflow obstruction. Typically, the left ventricular volume is normal or reduced. The left ventricular systolic function is usually hyperdynamic or normal but may be decreased in patients with a mixed HCM-DCM phenotype. There may be associated systolic anterior motion of the mitral valve with mitral regurgitation. HCM phenotype is a great masquerader and the echocardiographic impression of myocardial wall hypertrophy could be secondary to underlying cardiomyocyte hypertrophy, hyperplasia, interstitial edema, fibrosis or infiltrative/ storage disease. All of these histopathologic features give rise to the same phenotypic finding of hypertrophy on echocardiogram. The classic histopathologic features of sarcomeric HCM include myocyte hypertrophy, myofiber disarray, and patchy fibrosis. The most common mode of inheritance is autosomal-dominant, followed by matrilinear, X-linked, and autosomal-recessive inheritance patterns.
LVNC
LVNC11 is characterized by a “spongy” appearing LV with deep trabeculations and frequently associated with ventricular dilation and/or hypertrophy and impaired systolic and diastolic function. LVNC represents an arrest in the normal process of myocardial compaction, resulting in persistence of multiple prominent trabeculations and deep intertrabecular recesses. It typically involves the LV, although involvement of the RV has been reported. Lack of awareness of this disease may result in it being erroneously labelled as HCM or DCM. Autosomal dominant, X-linked, or mitochondrial inheritance patterns may be seen.
RCM
RCM12 is a distinct form of cardiomyopathy associated with predominant left ventricular or biventricular diastolic dysfunction in the setting of usually preserved systolic function. It can be primary or secondary in etiology. RCM is very rare in neonates, except for neonatal EFE, which may have a DCM, RCM or mixed DCM-RCM phenotype.
Etiologic Classification of Neonatal Cardiomyopathies
Neonatal cardiomyopathies have significant etiologic heterogeneity with multitude of genetic and acquired causes which have variable presentation and prognosis. A genetic basis can be identified in many cases of neonatal cardiomyopathies. Similar to infants and older children, genetic cardiomyopathies in neonates can be further subdivided into 4 major categories: (1) Inborn errors of metabolism (IEM); (2) Malformation syndromes (MFS); (3) Neonatal primary neuromuscular disorders (NMD); and (4) Familial isolated cardiomyopathy. IEM and MFS together account for one-third cases of infantile cardiomyopathy and likely a higher percentage of neonatal cardiomyopathy. The acquired cardiomyopathies could be due to infectious- or immune-related processes, maternal endocrine disorders, in-utero placental insult, perinatal asphyxia, coronary anomalies, fetal volume overload, fetal/neonatal paroxysmal tachyarrhythmias or iatrogenic drug induced.
Neonatal cardiomyopathies have significant etiologic heterogeneity with multitude of genetic and acquired causes which have variable presentation and prognosis. A genetic basis can be identified in many cases of neonatal cardiomyopathies. Similar to infants and older children, genetic cardiomyopathies in neonates can be further subdivided into 4 major categories: (1) Inborn errors of metabolism (IEM); (2) Malformation syndromes (MFS); (3) Neonatal primary neuromuscular disorders (NMD); and (4) Familial isolated cardiomyopathy. IEM and MFS together account for one-third cases of infantile cardiomyopathy and likely a higher percentage of neonatal cardiomyopathy. The acquired cardiomyopathies could be due to infectious- or immune-related processes, maternal endocrine disorders, in-utero placental insult, perinatal asphyxia, coronary anomalies, fetal volume overload, fetal/neonatal paroxysmal tachyarrhythmias or iatrogenic drug induced.
The common etiologic categories of neonatal cardiomyopathy are listed below.
Genetic Neonatal Cardiomyopathies
- Inborn errors of metabolism
- A. Disorders of cardiac energy metabolism
- Disorders of carnitine shuttle system (HCM, DCM)
- Disorders of long-chain fatty acid beta oxidation (HCM, DCM)
- Disorders of mitochondrial oxidative phosphorylation (HCM, DCM, LVNC)
- Disorders of carnitine shuttle system (HCM, DCM)
- Myocardial infiltration/storage disorders
- Glycogen storage diseases (HCM common, DCM rare)
- Lysosomal storage disorders (HCM common, DCM rare)
- Glycogen storage diseases (HCM common, DCM rare)
- Other miscellaneous IEM with cardiomyopathy
- Congenital disorders of glycosylation (HCM)
- Certain aminoacidurias and organic acidemias (DCM, HCM)
- Neonatal hemochromatosis (HCM, DCM)
- Peroxisomal disorders (HCM, DCM)
- Congenital disorders of glycosylation (HCM)
- A. Disorders of cardiac energy metabolism
- Malformation syndromes
- Neonatal Noonan syndrome and other ‘RASopathies’ (HCM)
- Neonatal overgrowth syndromes such as Beckwith-Weidemann, Simpson-Golabi Behmel syndrome (HCM)
- Chromosomal aneuploidies and deletion syndromes (DCM, LVNC, HCM)
- Neonatal Noonan syndrome and other ‘RASopathies’ (HCM)
- Neonatal Primary Neuromuscular disorders
- Congenital myopathies (DCM)
- Congenital muscular dystrophies (DCM)
- Congenital myopathies (DCM)
- Familial Isolated Cardiomyopathy secondary to genetic abnormalities of myocardial contractile and structural proteins (eg, dystrinopathies, laminopathies, sarcomeric familial HCM, usually compound/double heterozygosity). (HCM, DCM, LVNC)
Acquired Neonatal Cardiomyopathies
- Infectious
- Neonatal/ Perinatal Myocarditis (DCM)
- Sepsis Cardiomyopathy (DCM)
- Neonatal/ Perinatal Myocarditis (DCM)
- Hypoxic-Ischemic
- Perinatal asphyxia (DCM)
- Coronary artery stenosis (DCM)
- Anomalous Origin of Left Coronary Artery (ALCAPA) (DCM)
- In-utero coronary insult (DCM)
- Perinatal asphyxia (DCM)
- Fetal volume overload
- Twin-to-Twin Transfusion (HCM)
- Severe fetal anemia (DCM)
- Twin-to-Twin Transfusion (HCM)
- Fetal tachyarrhythmia
- Paroxysmal fetal SVT (DCM)
- Maternal Autoimmune Disease
- Neonatal lupus (DCM, EFE)
- Neonatal thyrotoxicosis (DCM)
- Neonatal lupus (DCM, EFE)
- Maternal Endocrine Disease
- Infant of diabetic mother (HCM)
- Drug Induced
- Steroid therapy (HCM)
The common individual disease entities of genetic and acquired neonatal cardiomyopathies are discussed later under the section of specific cardiomyopathies.
Epidemiology
Pediatric cardiomyopathies are rare disorders with an estimated incidence of 1.1 to 1.2 per 100,000 children with rates of 8 to 12 times higher in infants.13–15 In the postnatal period, cardiomyopathies account for 3% of neonates with cardiovascular diseases. There is paucity of epidemiologic data regarding neonatal and fetal cardiomyopathies. Based on data from limited case series, cardiomyopathies account for 8% to 11% of cardiovascular diagnoses detected in utero.16,17 Fetal cardiomyopathy is associated with high intrauterine loss in almost one-third of cases.16
DCM is the most common phenotypic form of pediatric cardiomyopathy, with an incidence of 0.57 to 0.58 per 100,000 children, and makes up more than 50% of the cardiomyopathies observed in the pediatric age group.18 Incidence is almost 8 to 10 times higher in infants, with a reported incidence of 4.4 per 100,000 infants in North America.19 Four in 10 cases of DCM in the North American Pediatric Cardiomyopathy Registry (PCMR) presented in infancy.19
HCM is the second most common type of cardiomyopathy in children and accounts for ~25% to 40% of pediatric cardiomyopathies with an incidence of 0.47 per 100,000 children.20 The annual incidence is much higher in infants and estimated to be ~3 per 100,000 infants.20 Infants account for almost one-third of pediatric cases of HCM. LVNC may account for ~9.5% of pediatric cardiomyopathies, and may be isolated or associated with CHDs (14%) or dysmorphism (14%).21
No gender based difference has been noted in the overall incidence of cardiomyopathies in infants, though there is a slight preponderance in males after one year of age.14 Cardiomyopathy due to metabolic causes is more common in males than in females.14,15,22 Higher incidence of cardiomyopathy has been reported in children of Black or Hispanic origin in North America and in indigenous children in Australia.14,15
In epidemiologic studies from North America, most children (~70%) with DCM did not have an established cause. Myocarditis accounted for 16%, neuromuscular disease for 9%, familial cardiomyopathy for 5%, IEM for 4% and MFS for 1% of the DCM cases. The high incidence of idiopathic classification was likely due to lack of comprehensive genetic and metabolic testing. Recent studies have shown that a comprehensive metabolic and genetic etiologic work up can decrease the percentage of idiopathic pediatric cardiomyopathies to almost 25%.23
Children with cardiomyopathy, secondary to IEM and MFS tend to present earlier and each account for ~15% of cases of infantile HCM in North America.20 Neuromuscular disorders on the other hand present later and account for only 1% of infantile diagnosis of HCM20 and 2% of infants with DCM.19 Almost 70% of infants with HCM fall in the category of idiopathic HCM.20 The causal identification in HCM is hampered by lack of metabolic and genetic investigation. In the PCMR, children with HCM who had undergone metabolic blood and urine testing were more likely to have a causal diagnosis compared to those who did not undergo metabolic screening (odds ratio 4.15).24 No gender preponderance has been noted in infantile HCM.
Clinical Presentation
Neonates with cardiomyopathy have a wide spectrum of clinical presentation, ranging from symptomatic congestive heart failure, multi-organ shock, hydrops fetalis, acute biochemical crisis, arrhythmia, encephalopathy, generalized muscle weakness/hypotonia, dysmorphic features, and sudden death to an asymptomatic neonate with an incidental diagnosis of cardiomyopathy upon echocardiographic evaluation for an unrelated illness or physical finding. Table 42.1 lists the association of antenatal maternal and fetal findings with specific cardiomyopathies.
| Finding | Cardiomyopathy subtype | Specific Cardiomyopathy |
|---|---|---|
| Antenatal Maternal History | ||
| Maternal diabetes (Type I, Type II, GDM) | HCM | Infant of diabetic mother (more common in Type I and Type II maternal DM) |
| Maternal thyroid diseases | ||
| 1. Graves’ | DCM | Neonatal thyrotoxicosis |
| 2. Hashimoto’s | DCM | Neonatal thyrotoxicosis (rare) |
| Maternal perinatal viral infection | DCM | Acute viral myocarditis (usually Coxsackie) |
| Maternal autoimmune disease SLE, AntiRho-AntiLa Abs | EFE, DCM, CAVB | Neonatal lupus |
| Maternal HELLP syndrome | HCM, DCM | |
| Antenatal Fetal History | ||
| Polyhydramnios | TTTS, lysosomal storage disorders, Beckwith-Wiedemann Syndrome | |
| Non-immune fetal hydrops | DCM, HCM | SVT-induced cardiomyopathy, lysosomal storage disorders, congenital disorders of glycosylation, TTTS |
| Fetal tachycardias | DCM | Tachycardia-induced DCM |
| IUGR | ||
| Fetal macrosomia | HCM | Beckwith-Wiedemann, Sotos, neonatal hyperinsulinism syndromes |
| Decreased fetal movements Arthyrogryposis multiplexa | DCM | Congenital myopathies, congenital muscular dystrophies |
| Monochorionic twins with significant growth disparity (≥20%)/fetal hydrops in one twin/polyhydramnios in one twin | HCM, DCM | Twin-to-twin transfusion syndrome |
| Intrapartum history | ||
| Fetal distress with non-reassuring fetal heart tones | ||
| Low APGARS | DCM | Perinatal asphyxia |
| Cord blood pH < 7.1 | DCM | Perinatal asphyxia |
| Family History | ||
| Cardiomyopathy, suspected AD inheritance | DCM, HCM, LVNC | Familial isolated cardiomyopathy |
| Cardiomyopathy, suspected matrilinear inheritance | DCM. HCM, LVNC | Mitochondrial cardiomyopathy |
| Consanguity | HCM, DCM, LVNC, EFE | Metabolic cardiomyopathies |
| Congenital pulmonary stenosis with poor school performance | HCM | Noonan’s syndrome |
| HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; LVNC, left ventricular non-compaction cardiomyopathy; | ||
| CAVB, complete atrioventricular block; EFE, endocardial fibroelastosis; TTTS, twin-to-twin transfusion syndrome. | ||
Cardiac Manifestation of Neonatal DCM
Almost 40% of children with DCM are diagnosed in infancy, and 70% of these infants have symptoms of heart failure at diagnosis.18,25 The echocardiographic evaluation is performed either due to signs and symptoms of congestive heart failure and low cardiac output or as part of a screening evaluation due to an asymptomatic murmur, dysmorphic features, or biochemical abnormality. The signs and symptoms depend on degree of ventricular dysfunction, the rate at which ventricular dysfunction develops and may range from asymptomatic ventricular dysfunction to overt CHF and cardiogenic shock. Heart failure is often progressive. Symptoms in infants primarily include tachypnea, poor feeding and failure to thrive. Signs and symptoms may be subtle at first and unmasked by a superimposed infectious illness that results in cardiac decompensation. Arrhythmias, thromboembolism, and sudden death are common and may occur at any stage. On examination, the most striking feature is tachycardia with decreased heart rate variability. Blood pressure is low to normal and is usually maintained until just before cardiovascular collapse. Cyanosis is uncommon. Jugular venous distension and peripheral edema is uncommon in infants. There is evidence of respiratory distress in patients with pulmonary edema. Infants may have “cardiac wheezing” due to airway obstruction caused by bronchial edema. There is usually cardiomegaly which may be associated with a prominent S3 and gallop rhythm and/or a murmur of mitral regurgitation in some patients. There may be associated hepato-splenomegaly in patients with CHF.
Cardiac Manifestation of Neonatal HCM
Neonatal HCM encompasses a heterogeneous group of diseases with diverse genetic origins and clinical phenotypes. Sarcomeric HCM is seldom the first entity in the differential diagnosis in a neonate. In neonatal/infantile HCM, the initial presentation may be an asymptomatic murmur (25%), symptomatic congestive heart failure, or murmur and/or arrhythmia associated with symptoms of tachypnea, tachycardia, and poor feeding. Compared to older children with HCM, infants with HCM are more likely to be symptomatic and almost one-third are symptomatic with CHF at presentation.
Extracardiac Manifestations of Neonatal Cardiomyopathies
Neonates with cardiomyopathy, secondary to inborn errors of metabolism involving impaired energy production or toxic metabolite accumulation often have signs and symptoms of multiple organ dysfunction. Under conditions of energy stress (infection, starvation) they may present with hypoglycemia, metabolic acidosis, and/or hyperammonemia. Presence of unexplained acute or chronic encephalopathy, muscle weakness, hypotonia, growth retardation, failure to thrive, recurrent vomiting, and lethargy in a neonate with cardiomyopathy should result in screening for an underlying biochemical abnormality of energy metabolism. In contrast, neonates/infants with lysosomal/storage disorders who cannot degrade certain components of cells typically develop coarse or dysmorphic facial features, organomegaly, skeletal abnormalities, short stature or chronic encephalopathy associated with a neurodegenerative course. Dysmorphic features may be secondary to malformation syndromes as well as storage disorders. Skeletal muscle weakness without encephalopathy may indicate a primary neuromuscular disorder or disorder of energy metabolism. The clinical presentation of fatty acid oxidation disorders or respiratory enzyme deficiencies as cardiac dysfunction can be confusing and masquerade as acute myocarditis or isolated cardiomyopathy.
Imaging and Laboratory Findings
Chest Radiography
In patients with DCM phenotype, chest x-ray (CXR) may show cardiomegaly with various degrees of pulmonary venous congestion, including cephalization of pulmonary vascularity, interstitial edema, Kerley-B lines, pulmonary edema and pleural effusion. In addition to cardiomegaly, cardiac silhouette is usually abnormal with cardiac apex shifted outward and downward (suggestive of left ventricular dilation). There may be associated segmental collapse of left lower lobe due to compression of the left lower lobe bronchus by a dilated left atrium. Similarly, CXR in neonates with HCM may show cardiomegaly with outwardly shifted cardiac apex. There may be evidence of pulmonary venous congestion in the presence of significant left ventricular diastolic dysfunction. In the absence of significant CHF or cardiomegaly, CXR findings may be normal.
Electrocardiography
Electrocardiogram (ECG) abnormalities in neonates with cardiomyopathy are common and usually nonspecific. There may be evidence of chamber hypertrophy/enlargement and nonspecific ST-T segment and T-wave abnormalities. Pompe disease is characteristically associated with short PR interval with huge QRS voltages. Ventricular pre-excitation may be seen in Pompe disease, PRKAG2 mutations, lysosomal storage disorders and mitochondrial diseases. Patients with cardiomyopathy secondary to neonatal lupus usually have coexisting complete atrioventricular block (CAVB). Atrioventricular block may also be seen in isolated familial cardiomyopathies secondary to laminopathies, SCN5A mutations, as well as mitochondrial cardiomyopathies, Pompe disease and lysosomal storage diseases. Huge QRS voltages in septal leads may be seen in LVNC. Repolarization abnormalities are common but nonspecific findings, including ST segment depression and T-wave inversion. Infarction/pseudo-infarction pattern including abnormal Q waves, ST elevation, T-wave inversion with discordant QT vector may be seen in perinatal asphyxia, myocarditis, ischemic cardiomyopathy as well as HCM. Holter monitoring may reveal atrial or ventricular arrhythmias.26 Table 42.2 lists some ECG findings which are associated with specific neonatal cardiomyopathies. Figure 42.2, shows some characteristic ECG images.
Table 42.2. ECG findings which may point towards a specific diagnosis in neonatal cardiomyopathy
| Finding | Cardiomyopathy subtype | Specific Cardiomyopathy |
|---|---|---|
| Short PR interval with huge QRS voltages | HCM | Pompe disease |
| Ventricular Pre-excitation (WPW syndrome) | HCM | Pompe disease, PRKAG2 mutations, lysosomal storage disorders, mitochondrial disorders (MELAS, MERFF) |
| AV Block | DCM, EFE | Neonatal lupus (Anti-Rho/Anti-La) laminopathy, SCN5A mutation, myocarditis |
| AV Block | HCM | Mitochondrial cardiomyopathies, glycogen storage disease, Pompe), lysosomal storage disorders |
| Extremely High QRS voltage | HCM | Pompe disease, Danon disease, CM with subtle preexcitation |
| Low QRS voltage | DCM | Myocarditis |
| Infarction/pseudo-infarction pattern (Q wave >3.5 mm, usually in left-sided leads, ST elevation >2 mm) | DCM | Perinatal asphyxia, myocarditis, ischemic cardiomyopathy (coronary stenosis, large coronary fistula, rarely ALCAPA, in-utero thromboembolic phenomenon) |
| HCM:, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; AV Block, atrioventricular block; EFE, endocardial fibroelastosis. | ||
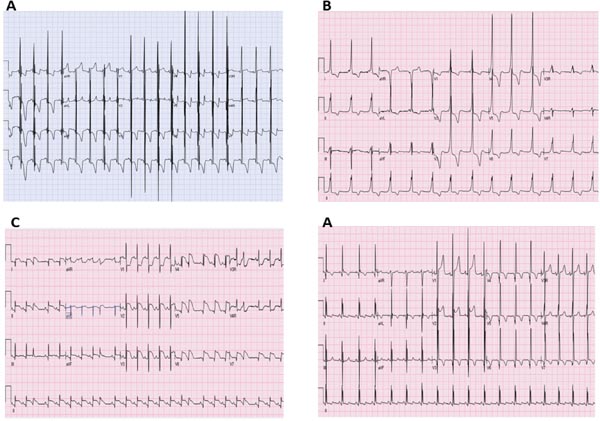
Figure 42.2. Some characteristic disease-specific ECGs: A: Pompe’s disease with short PR interval and huge QRS voltages. B: HCM with WPW syndrome secondary to PRKAG2 mutation. C: Pseudoinfarct like pattern in neonatal myocarditis with diffuse ST segment elevation and occasional premature ventricular contractions. D: Abnormal Q waves in left-sided leads (Leads I, AVL, V6) in an infant with ischemic cardiomyopathy secondary to Anomalous Left Coronary Artery from Pulmonary Artery (ALCAPA).
Echocardiography
Echocardiography in patients with DCM shows usually left and occasionally biventricular enlargement with global ventricular dysfunction. The left ventricular dimensions and function may be compared to body surface area based norms reported by Colan et al.27 Regional wall motion abnormality is less common and, if seen, should trigger evaluation for an underlying coronary abnormality. Anomalous origin of the left coronary artery and lesions causing left ventricular outflow obstruction need to be ruled out by 2D echocardiogram, especially in infants presenting with left ventricular dysfunction. Echocardiogram is also useful in detecting atrial or ventricular thrombi which can develop in the presence of low output state or atrial arrhythmias. There may be coexisting diastolic dysfunction.
Echocardiogram in neonates with HCM shows generalized or regional hypertrophy of the LV. This is determined by 2D or M-Mode echocardiographic measurements of the left ventricular posterior wall or interventricular septum in diastole, with any measurement ≥2 SDS (standard deviation scores) above the reported normal values for body surface area27,28 considered to be in the hypertrophic range. The hypertrophy may be concentric or asymmetric septal hypertrophy may be seen. Systolic function is usually hyperdynamic, but may be depressed especially in patients with storage/mitochondrial disorders. Coexisting diastolic dysfunction is common. Left ventricular cavity size is usually relatively small, but may be dilated in cases with mixed phenotype. In patients with asymmetric septal hypertrophy there may be coexisting dynamic left ventricular outflow obstruction and/or systolic anterior motion of mitral valve resulting in mitral regurgitation. Echocardiographic findings of neonatal cardiomyopathies are listed in Table 42.3 and some examples are shown in Figure 42.3.
Table 42.3. Echocardiographic findings of neonatal cardiomyopathy
| Echocardiographic Finding | Morphologic Phenotype |
|---|---|
| Fractional Shortening/Ejection Fraction >2 SD below normal mean for age | DCM |
| Left ventricular end diastolic dimension >2 SD above the normal mean for age | DCM |
| LV posterior wall thickness >2 SD above normal mean for body surface area | HCM |
| Interventricular septal wall thickness > 2SD above normal for body surface area | HCM |
| Isolated asymmetric interventricular septal hypertrophy | HCM |
| Mitral regurgitation with left ventricular dilation | DCM |
| Mitral regurgitation with SAM | HCM |
| Trabeculated LV with focal/diffuse trabeculations with noncompacted to compacted zone >1.4:1 | LVNC |
| Dynamic LVOTO | HCM |
| Parchment-like right ventricle | Uhl syndrome |
| Echo bright endocardium with systolic and/or diastolic impairment | EFE |
| Primary ventricular diastolic dysfunction | Primary RCM |
| HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; LVNC, left ventricular noncompaction; RCM, restrictive cardiomyopathy; EFE, endocardial fibroelastosis; SD, standard deviation; LVOTO, left ventricular outflow tract obstruction; SAM, systolic anterior motion. | |
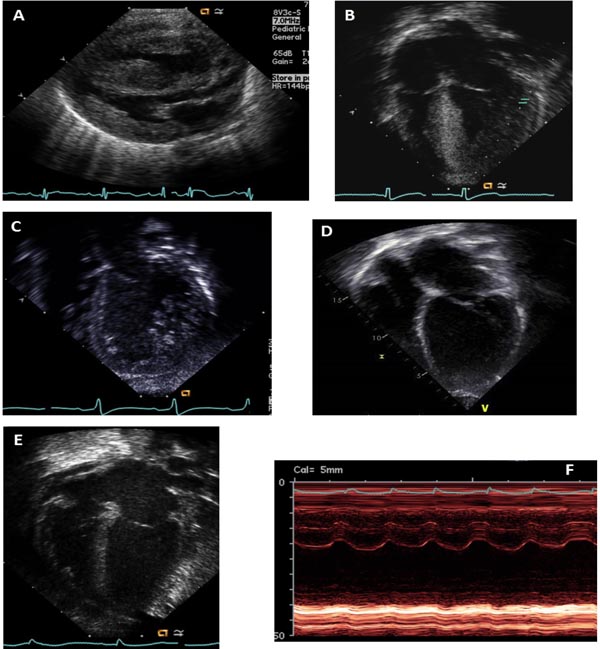
Figure 42.3. Echocardiographic findings in neonatal cardiomyopathies: A: Hypertrophic cardiomyopathy with asymmetric septal hypertrophy in an infant of diabetic mother. B: Hypertrophic cardiomyopathy with concentric left ventricular hypertrophy in a neonate with Pompe disease. C: Left ventricular noncompaction with a dilated and trabeculated left ventricle in a neonate with mitochondrial cardiomyopathy. D: Dilated and globular left ventricle in a neonate with idiopathic DCM. E: Mild left ventricular dilation. F: Severely depressed contractility of left ventricular free wall in a neonate with Coxsackie B myocarditis.
Other Cardiac Imaging Studies
Cardiac catheterization with endomyocardial biopsy may provide useful diagnostic and prognostic information, but is limited in neonates by its inherent risks and lack of sensitivity. It is performed as part of a pre-transplant evaluation to assess cardiac hemodynamics and pulmonary vascular resistance. Cardiac magnetic resonance imaging (MRI) can provide an accurate estimation of right and left ventricular volume and ejection fraction. It can be particularly helpful in distinguishing primary DCM from secondary causes of DCM like ischemic cardiomyopathy or acute myocarditis. Endomyocardial biopsy may be helpful in some cases such as neonatal myocarditis or cardiac phosphorylase kinase deficiency, a rare metabolic cause of infantile cardiomyopathy, but is limited by lack of procedural expertise in noncardiac transplant centers and concerns for higher complication rate in neonates. Endomyocardial biopsy in neonates should be performed only if the results will alter management of the underlying disease or significantly alter prognosis.
Biochemical and Serologic Evaluation
Besides the standard cardiac evaluation as above, the initial evaluation of pediatric DCM patients should include serologic biomarkers for myocardial injury or inflammation such as cardiac troponin T or I and CK MB (to rule out an underlying ischemic or inflammatory etiology), and blood and urine tests to rule out inborn errors of metabolism (serum amino acids, urine organic acids), mitochondrial dysfunction (serum lactate and pyruvate, acyl carnitine profile), or thyroid dysfunction. In cardiomyopathies due to disorders of fatty acid metabolism biochemical abnormalities including hypoketotic hypoglycemia, hyperammonemia, elevated creatine kinase and transaminases, dicarboxylic aciduria, very low free carnitine and abnormal acylcarnitine profile with marked elevation of the long-chain acylcarnitines is seen in symptomatic neonates.29 Patients with neonatal mitochondrial cardiomyopathies show a disproportionate lactic acidosis, usually accompanied by other associated metabolic alterations including hypoglycemia, hyperammonemia, ketonuria, and altered amino acid and organic acid chromatographic patterns (mostly increased alanine, proline, and glutamine, and accumulation of Krebs cycle metabolites). In selected patients suspected of a mitochondrial dysfunction, skeletal muscle biopsy should be performed with evaluation by light and electron microscopy and mitochondrial respiratory chain analysis. Serology and polymerase chain reaction (PCR) on tracheal aspirate and other body fluids for cardiotropic viruses should be performed in patients suspected to have viral myocarditis. However, the yield may be low and viral serologic studies have a low sensitivity in diagnosing acute myocarditis. Patients with an elevated erythrocyte sedimentation rate should undergo evaluation for a primary inflammatory or autoimmune process. Serum levels of B-type natriuretic peptide (BNP) should be measured at presentation and at serial follow up. The degree of BNP elevation in the setting of heart failure has been shown to be directly related to morbidity and mortality30 and may also be helpful in titrating therapy.
Diagnostic Approach to Neonatal Cardiomyopathies Based on Clinical Presentation
All neonates with suspected cardiomyopathy should undergo a systematic evaluation to identify an underlying ‘genetic’ or ‘acquired/mixed’ cause. Cause specific diagnosis is important for determination of prognosis and institution of cause specific therapy. The work up should include screening for biochemical metabolic abnormalities, assessment for extra-cardiac manifestations and presence of dysmorphic features and a comprehensive family history involving three generation pedigree with appropriate imaging.31 Possible acquired etiologies need to be considered based on the perinatal history.
Neonatal Cardiomyopathy with Biochemical Abnormalities
At initial presentation, all neonates with suspected/confirmed primary cardiomyopathy should undergo a measurement of blood glucose, acid-base status and biochemical markers of hepatic and renal function. The initial laboratory studies are aimed at determining the metabolic pathway which is the most likely site of abnormal biochemical defect. Presence of hypoglycemia, primary metabolic acidosis with an increased anion gap, or hyperammonemia is an indication to pursue further screening evaluation for an underlying inborn error of metabolism. Further diagnostic algorithms, based on the initial abnormal biochemical or physical findings, are available to aid in further diagnostic work up.32 Biochemical laboratory tests have maximum diagnostic yield when performed during acute phase of illness. Hence, blood and urine samples should be obtained as soon as possible after presentation. Additional plasma and urine should be frozen for further specialized testing. Table 42.4 lists the common laboratory tests for screening for presence of metabolic abnormalities in neonatal cardiomyopathies.
Table 42.4. Screening laboratory tests for metabolic evaluation of neonatal cardiomyopathies
| Plasma |
|---|
| Lactate Pyruvate Free fatty acids Ketone bodies Beta hydroxybutyrate Acetoacetate Total and free carnitine Acyl carnitine esters Ammonia Cholesterol Creatine kinase Amino acids |
| Urine |
| Organic acids Glycosoaminoglycans and oligosaccharides Glucose tetrasaccharide |
Associated hypoglycemia Significantly decreased oral intake in a neonate normally results in hypoglycemia with associated ketotic response. On the other hand, hypoketotic hypoglycemia is an abnormal response and considered the hallmark of an underlying fatty acid metabolism defect. Similarly, hyperketotic hypoglycemia is characteristic of defects in organic acid metabolism. Presence of ketones can also be a nonspecific finding in association with high lactate level as in patients with mitochondrial disorders presenting with acute metabolic crises.
Associated anion gap metabolic acidosis In neonates with cardiomyopathy and metabolic acidosis with an increased anion gap, identification of the organic acid(s) in blood and/or urine which is responsible for the increased anion gap is the key to the diagnosis. These commonly include elevated free fatty acids, ketoacids, dicarboxylic acids, lactate and pyruvate. Once a diagnosis is suspected based on the abnormal array of biochemical tests, the specific diagnosis should be confirmed by enzyme assay or increasingly available deoxyribonucleic acid (DNA) mutation analysis. Plasma acylcarnitine profile and urinary acylglycine analysis can detect low levels of abnormal diagnostic metabolites in asymptomatic patients who otherwise lack any gross detectable biochemical changes in blood and urine.
Neonatal Cardiomyopathy with Encephalopathy
In a neonate with cardiomyopathy, coexisting encephalopathy in the form of impaired mental status, coma, seizures, central apnea, autonomic dysfunction, dystonia or stroke-like episodes, has important diagnostic significance. Acute encephalopathy is frequently associated with metabolic decompensation and should trigger an investigation for an underlying biochemical abnormality. Chronic encephalopathy is characteristic of mitochondrial syndromes such as MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes), MERRF (Myoclonic Epilepsy, Ragged Red Fibers), Kearn-Sayre syndrome, and Leigh disease. An intercurrent illness or metabolic stress may precipitate an acute worsening in these syndromes. Hence diagnostic work up for cardiomyopathies with coexisting neuro-encephalopathic manifestations such as seizures, stroke-like episodes, neurocognitive developmental delay or opthalmoplegia should include brain MRI, measurement of lactate and pyruvate levels in blood and cerebrospinal fluid, skeletal muscle biopsy for light and electron microscopic histology. It also should include functional assay of respiratory chain enzyme activity level and genetic analysis for mitochondrial DNA or mitochondrial protein encoding nuclear DNA mutations. Loss of neurodevelopmental milestones associated with valvar heart disease, coarse facial features, organomegaly, skeletal deformities, or cloudy corneas may suggest the presence of an underlying lysosomal storage disorder such as mucopolysaccharidosis or mucolipidosis.
Neonatal Cardiomyopathy with Hypotonia and Muscle Weakness
Neonatal presentation of cardiomyopathies with associated neuromuscular disorder is rare. Neuromuscular disorders account for only 1% of cardiomyopathies presenting in infancy. In the rare cases that present in the neonatal period/infancy, the neurologic symptoms may include congenital hypotonia, muscle weakness beginning in infancy or congenital myotonia. When cardiomyopathy is associated with hypotonia or weakness in a neonate or older infant, an IEM is more likely than a primary congenital neuromuscular disorder. Specific neuromuscular disorders presenting in the neonatal/infantile period include congenital myopathy, congenital muscular dystrophy and congenital myotonic dystrophy. At initial presentation, the primary manifestation is neuromuscular and cardiomyopathy usually develops later. Findings which support the diagnosis of a neuromuscular disorder include a distinct pattern of muscle weakness, decreased muscle bulk, decreased or absent deep tendon reflexes, elevated serum creatine kinase levels, and an abnormal nerve conduction velocity or electromyogram. It is important to realize that nerve conduction velocity and electromyography are difficult to interpret in neonates/young infants and mild disease may not be detected when performed early. A muscle biopsy and/or disease specific genetic mutation analysis is required to establish an underlying definitive diagnosis.
Neonatal Cardiomyopathy with Dysmorphism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


