ANGIOGENIC SWITCH IN CANCER: WHOLE-ORGAN STUDIES
A milestone of molecular biology and cancer research is the development of specific gene silencing and upregulation techniques in in vivo mouse models. Several mouse models of the angiogenic switch are described in the succeeding discussion.
Mouse Models The RIP-Tag mouse model was created by genetically manipulating mouse pancreatic
β-cells to express the SV40 large T antigen; a hyperproliferative stage arises in about half the pancreatic islets, and in a minority of those, pancreatic islet cell carcinoma develops. The induction of angiogenic activity demarcates an angiogenic stage, which is found in islets that have progressed past the proliferative stage and apparently precedes carcinoma.
7 The isolation and examination of these islets revealed that an angiogenic phenotype is not a direct outcome of oncogene expression, nor is it the inevitable effect of a proliferative mass that requires an increased blood supply. The RIPTag model demonstrated discrete stages of tumor progression in which additional genetic manipulation can be used to determine the roles of various molecules in each phase. Thus, this model was used to evaluate the roles of vascular endothelial growth factor (VEGF) in the angiogenic switch,
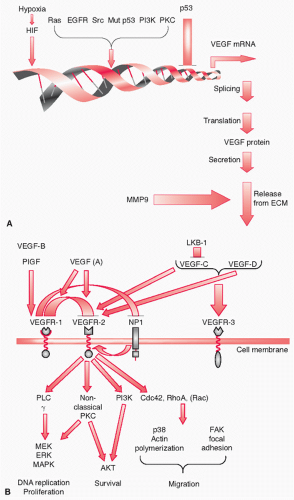
8 matrix metalloproteases (MMP9) in the release of VEGF from the extracellular matrix (ECM),
9 and VEGF receptor 2 (VEGFR-2) expression on vascular endothelial cells
10 (see succeeding discussion).
Tumor fragments transplanted onto the irises of rabbits were used in a classic model that demonstrated the importance of the angiogenic switch.
11 In some cases, the tumor fragments induced angiogenesis and progressed; in others, they remained clinically dormant. Active proliferation and apoptosis were found at the cellular level in clinically dormant tumors. Similar apparent dormancy has also been described in other conditions in which angiogenesis was inhibited.
12The angiogenic switch is required not only for the progression of primary tumors but also for the growth of metastatic deposits. In a mouse model of metastatic disease, removal of the primary tumor induced rapid growth in previously dormant metastatic deposits. An angiogenesis inhibitor produced by the primary tumor was later identified. The rapid growth of the micrometastasis was accompanied by angiogenesis induction and a concomitant apoptosis reduction, with no change in the proliferative rate.
12 Both activators and inhibitors of angiogenesis are produced in intact organisms, and the balance between them determines the vascularity of the tumor and its metastasis and progression.
Angiogenesis Evaluation in Human Cancer In clinical studies, the most useful method for evaluating tumor vascularity is counting microvessel density (MVD) in tumor sections by immunohistochemically staining for one of several molecular markers of endothelial cells, namely factor VIII, von Willebrand factor, CD31, or CD34. Under low magnification, areas of high MVD can be observed, usually at the tumor periphery. The actual MVD is determined in those areas by counting the number of capillaries per high-power field. Various protocols for counting or subjective visual evaluation exist.
13 MVD has been correlated with imaging-determined tumor perfusion
14 and clinical outcome in many tumor types. However, many vessels visualized by immunohistochemical staining may not be functional. CD 105 staining was recently suggested to be specific for active blood vessels as opposed to general endothelial markers that stain also nonfunctional vessels.
15 Proliferating endothelial cells can also be assessed by immunohistochemical analysis, because they are a more reliable marker of ongoing angiogenesis than MVD. Double-staining for endothelial-specific proteins and Ki-67 is used to estimate the proportion of proliferating cells; this method was better correlated with stage than counting MVD in colorectal cancer.
16 An important caveat of these methods is the high variability within tumors
17; thus, sampling errors may be significant. Although highly useful and clinically prognostic in many studies (see later), these methods
provide no information about tumor perfusion, which is the biologically relevant end point of the angiogenic switch. For this, other approaches must be used.
An indirect method of evaluating tumor angiogenesis is the assessment of tumor hypoxia. Carbonic anhydrase IX, a transcriptional target of hypoxia-inducible factor 1 (HIF-1), may be a surrogate marker of tumor hypoxia. High levels are found in hypoxic areas in several cancer types, including NSCLC, and its expression is associated with a poor prognosis.
18 Pimonidazole is an exogenous marker of hypoxia
19 that can be used in immunohistochemical analyses of biopsy samples
20; however, this method is not commonly used, because it requires pimonidazole to be intravenously infused to patients prior to biopsy. Interestingly, tumor cell necrosis, a plausible indicator of tumor hypoxia, has not been reported to be a prognostic factor in lung cancer. Nuclear medicine allows molecular imaging, including the ability to detect hypoxia. The positron emission tomography tracer 18F-misonidazole
21 and other tracers are being assessed as prognostic or predictive markers in several cancer types. However, such tools require further validation and are not yet available in most cancer centers.
Major inducers and inhibitors of angiogenesis can be used as surrogate markers for angiogenesis in cancer. The levels of VEGF, its receptors, and various endogenous facilitators and inhibitors of angiogenesis can be evaluated in patients’ tumor tissues or serum. Importantly, such studies can be performed in large-scale setups and may be useful clinically. Alternatively, immunohistochemical analyses of protein expression in tumors can be performed, but these have the same potential sampling bias of MVD studies.
Additional parameters of angiogenic activity that can be assessed from a blood sample include circulating endothelial cells (CECs) and circulating endothelial progenitor cells (circulating EPC; CEPs). CECs were first reported more than 30 years ago by Hladovec and Rossmann.
22,
23 Mature CECs probably originate from cells shed from vessel walls. Some of such cells possess progenitor characteristics and are referred to as CEPs; they are thought to originate from the bone marrow. Both CECs and CEPs may be useful surrogate markers for angiogenic activity and for tumors’ response to antiangiogenic treatment.
24 However, the assessment of CECs or CEPs in patients’ blood is technically challenging, and no consensus exists about assessment methods or even CEPs’ significance (see discussion later).
Tumor perfusion can be assessed in vivo using various imaging studies. Most of these methods are investigational, although promising. Microscopic bubbles can be used as contrast material in sonography studies to demonstrate blood flow in vivo. Computed tomography and magnetic resonance imaging using intravenous contrast material can also be used to measure blood flow and volume. Positron emission tomography nuclear imaging using
11C- or
15O-marked carbon monoxide can be used for the same purpose. Molecular imaging, in which the contrast material is conjugated to a molecule that binds to a cancer endothelialspecific epitope, is currently being evaluated.
25Angiogenic Switch in Human Cancer Stepwise progression is evident in several cancer types, and discrete stages can be differentiated in pathologic specimens. For example, breast cancer is preceded by carcinoma in situ, in which vessel density is correlated with several poor prognostic factors.
26 Additional indicators of angiogenic activity, such as messenger RNA (mRNA) expression levels of VEGF and its receptors, were upregulated in breast in situ carcinoma, similar to what was found in invasive breast cancer.
27 Cervical squamous cell intraepithelial neoplasia is the precursor of cervical carcinoma and is graded according to the proportion of dysplasia. Vessel density in the stroma below the basement membrane of the dysplastic epithelium was correlated with the grade of epithelial dysplasia.
28 Evaluation of dysplastic bronchial epithelium, lung premalignant lesions, revealed increased MVD and increased levels of VEGF levels compared with normal controls. A characteristic pattern of VEGFR and VEGF isoform expression comparable to invasive lung cancer was found. Apparently normal lungs of heavy smokers harbored enhanced VEGF mRNA levels.
29 Abnormal microvasculature structure was described to appear near dysplastic squamous bronchial epithelium.
30 The results of these studies suggest that, similar to what was demonstrated in mouse models, human cancer must acquire a vascular supply in order to progress. At least in some types of cancer, including lung cancer, the angiogenic switch occurs prior to the invasive phase of cancer progression.
Angiogenic Signaling in Response to Hypoperfusion Oncogene activation and tumor suppressor gene inactivation can activate the angiogenesis switch (see later). However, hypoperfusion is an alternative, and conceptually antagonizing, source of proangiogenic signals. Tumors that outgrow their blood supply experience reduced oxygen and glucose levels. Hypoxic regions are commonly found in several cancer types. These regions may indicate highly dysregulated cell growth; they are associated with a poor prognosis.
31,
32 Hypoxia may select for more aggressive tumor cells
33 or activate signaling pathways that lead to increased invasion and metastasis. For example, in NSCLC cells, hypoxia induced in a HIF-1
β-dependent manner CXCR4 expression.
34 CXCR4 is a cytokine receptor involved in invasion and metastasis.
35 A direct consequence of hypoxia and hypoglycemia is HIF-dependent induction of VEGF expression in malignant cells.
36 VEGF activates an angiogenic response, which results in new blood vessels, improved tumor vascularization, and potentially relief of hypoxia and hypoglycemia. VEGF induction by hypoxia or hypoglycemia involves an apparently normal feedback mechanism, in which reduced perfusion activates a corrective mechanism. Normally, once perfusion has improved, VEGF secretion is reduced.
36 Hypoperfusion-induced signaling is not perpetual, unlike the angiogenic switch. However, newly formed blood vessels of tumors are not as functional as mature vessels of normal tissues: They are only partially covered by pericytes and are highly permeable, torturous, and chaotic.
25 The inefficiency of the newly formed tumor blood supply causes the hypoxia-induced signaling to prevail and paradoxically contributes to cancerous growth.
CELLULAR PLAYERS AND MECHANISMS IN TUMOR VASCULARIZATION
Endothelial Sprouting and Pericyte Coverage Tumor blood vessels form and develop with tumors by several mechanisms, the most studied of which is endothelial sprouting, whereby new capillaries bud from nearby existing ones. Sprouting can proceed through a phase of a vascular network that is superfluous and undergoes pruning, or it can be guided to hypoperfused regions as it is created, mostly by VEGF gradients.
37 The phases of endothelial sprouting have been carefully described.
38 The basement membrane of postcapillary venules is degraded at the location of the future endothelial sprout. Through the resultant opening in the basement membrane, endothelial cells migrate and form a cord of cells; this is followed by the appearance of a lumen. According to another report, the sprouting vessel sustains a lumen and continuous intracellular junctions as it is produced, rather than going through a stage of dedifferentiated cord of cells as commonly thought.
39
Endothelial cell migration is a major process in angiogenesis. It is regulated by chemotaxis toward VEGF, basic fibroblast growth factor (bFGF), and other angiogenic factors. It is also controlled by haptotaxis, the migration toward a gradient of immobilized ligands, which is dependent on integrin-ECM interactions. Mechanotaxis is the sensing of sheer stress of blood flow by cytoskeletal elements and migration in its direction and is another mechanism that controls endothelial migration.
40,
41The last phase of the angiogenesis process is the recruitment of pericytes and deposition of new basement membrane.
42 Pericytes of the parent blood vessel proliferate and migrate to envelop the new vessel.
39 The platelet-derived growth factor (PDGF) pathway is the major regulator of pericyte recruitment and maintenance. PDGF-B is secreted mostly by endothelial cells. Acting on PDGF receptor-
β (PDGFR-
β) on pericytes, it facilitates their recruitment to new blood vessels. Pericytes, in turn, contribute to the stability and functionality of blood vessels, partly through the secretion of VEGF.
43 Importantly, tumor blood vessels that lack pericyte coverage are the first to regress after VEGF pathway inhibition.
44 Tumor blood vessels contain more than one subtype of pericytes, which vary in their molecular marker expression and dependence on PDGF signaling for tight adhesion to endothelial cells.
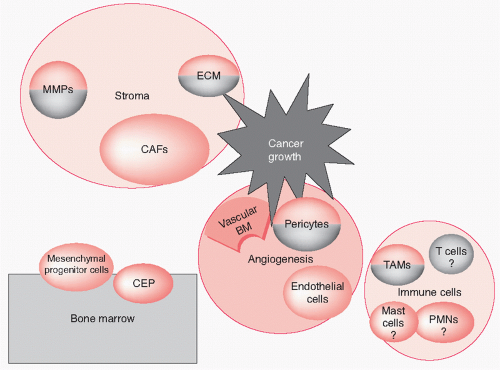
45The origin of tumor vascular pericytes is thought to be mesenchymal progenitor cells,
43 which are characterized by Tie2 expression,
46 or bone marrow-derived hematopoietic stem cells.
47 Some models indicate that endothelial cells and pericytes share a common angioblast progenitor cell.
48 The differences between vascular smooth muscle cells and pericytes are not clear, suggesting that they are similar cell types in different phases of phenotypic change.
3 Regardless of the origin of these cells, the clinical activity of PDGFR inhibition in the treatment of cancer indicates that pericytes are important in the maturation and modulation of tumor angiogenesis
43 (see
Fig. 8.1 for a schematic representation of the major cell types and niches that modulate tumor angiogenesis).
Vasculogenesis: Cells from the Bone Marrow Vasculogenesis is the de novo formation of blood vessels from vascular progenitor cells. Circulating bone marrow-originating cells travel to specific foci and undergo in situ differentiation
to form mature components of blood vessels. Vasculogenesis was initially thought to occur only in embryonic tissues, but it has been found to occur in adults as well. Bone marrow-derived endothelial progenitor cells (EPC) were found to be mobilized (thus becoming CEPs) by GM-CSFs or ischemia in experimental animals and travel to areas of ischemia.
49 Studies in which mice underwent bone marrow transplantation from mice that expressed a unique marker demonstrated that bone marrow-derived cells contribute directly to blood vessel formation.
49,
50 VEGFR-2 is critical for vasculogenesis in embryos
51 and adult tissues.
52 In addition, increased expression of stromal cell-derived factor-1 (SDF-1, also called CXCL12)-in peripheral blood and ischemic tissues, in parallel to reduced SDF-1/CXCL12 expression in bone marrow, may enhance the recruitment of CEPs to ischemic tissues. SDF-1/CXCL12 enhanced the number of EPC in ischemic vessels by promoting their adhesion through
α2,
α4, and
α5 integrins to fibronectin and collagen I.
53 CEPs have also been found in patients, in numbers that were correlated with plasma levels of VEGF 165.
54
CEPs were also shown to contribute to the formation of blood vessels in cancer. Id knockout mice display defective angiogenesis, not allowing them to support the growth of implanted tumors.
55 This phenotype was saved by transplantation with wild-type bone marrow. Donor-derived, VEGFR-2-positive CEPs formed most of the tumor blood vessels in this model. Donor-derived, VEGFR-1-positive myeloid precursors were also recruited to the tumors, where they were thought to have secreted angiogenic cytokines.
52 The role of inflammatory cells in angiogenesis will be discussed later. The expression of the transcription factor Id1 in CEPs was essential for their contribution to lung metastasis in another mouse model.
56 Systemic 17-
β estradiol administration contributed to the recruitment of CEPs to tumors in a mouse model of breast cancer.
57 Blood counts of CEPs may be a promising surrogate marker for angiogenesis or vasculogenesis, possibly useful in the real-time assessment of the efficiency of treatment targeting tumor blood vessels.
24 For example, vascular-disrupting agents induced a surge in the CEP blood concentration in a mouse cancer model. Treatment with anti-VEGFR-2 antibody disrupted this surge and augmented the efficacy of cancer eradication. Blocking the CEP surge prevented the regrowth of tumor from the rim of viable cells that typically remain when most of the tumor necrotizes.
58 Cancer-associated fibroblasts (CAFs), a prominent component of the stromal reaction to cancer, contributed to the recruitment of CEPs.
59 SDF-1/CXCL12 release by CAFs is critical to this recruitment.
59 CEPs were found in the blood of cancer patients and were demonstrated to respond to effective systemic therapy.
60 Therefore, bone marrow-derived EPCs may be one of the important manners by which tumors develop their vascular supply.
The importance of EPCs is controversial, spurring disputes in the scientific literature.
61,
62 Estimations of their contributions to tumor endothelium vary from significant (10% to 50%) to negligible.
24,
63 Studies finding no evidence of EPC contribution to tumor endothelium were also reported.
61 A plausible explanation for this variability was suggested when time-dependent changes in EPC contribution were evaluated. Using high-resolution microscopy, aided by flow cytometry, bone marrow-originating cells can be located in inoculated tumors in mice that have undergone bone marrow transplantations of GFP-positive cells. In this model, the proportion of EPCs among endothelial cells was about 30% in the first 4 to 6 days of tumor growth but dropped to less than 1% after 4 weeks. This study also demonstrated bone marrow-derived cells close to endothelial cells, suggesting that they are a source of EPC overestimation in tumor vessels.
64 A study of cancers that developed in bone marrow transplant recipients that are gender mismatched with their donor revealed that about 5% of their tumor endothelial cells are donor-originated.
63 Importantly, almost all of the CEC that had a significant proliferative capacity, were donor-originated.
65 This study suggests that even very low numbers of EPC might contribute significantly to tumor vasculature.
The evaluation of CEPs in blood samples of cancer patients is hampered by the low numbers of these cells in circulation and the technical difficulties of their positive identification. Identification methods include enrichment by cell sorting and immunomagnetic beads. However, these methods depend on specific surface markers, which are lacking. For example, CD 146 may be a specific marker of CEPs or CECs and may be measurable in the serum of cancer patients. CD146 mRNA levels in serum were correlated with CECs in breast cancer patients.
66 However, a fluorescence-activated cell sorting analysis of blood mononuclear cells demonstrated that CD146 was expressed mainly on a subpopulation of T cells.
67 Recently, tumor endothelial marker 1/endosialin/CD248 was reported to be highly expressed in CEPs, suggesting a new method of measuring or potentially eradicating blood CEPs.
68 An important property of CEPs that may be useful in their identification is their ability to proliferate, but this would not differentiate CEPs from hematopoietic progenitor cells. The technical difficulties of detecting a minute subpopulation of cells in the blood have not been satisfactorily solved.
24 The results of studies of CEPs and CECs in the blood of cancer patients must be interpreted cautiously.
Alternative Mechanisms of Enhanced Vascularization In vessel co-option, tumors or metastatic foci develop along existing blood vessels. In this way, tumors become vascularized with no need for blood vessel formation.
69 Vessel co-option occurs in the initial growth phase of tumors. As cancer cells proliferate, the tumor outgrows its blood supply. The co-opted host blood vessels then undergo regression, possibly as a host defense mechanism. The endothelial cells of these vessels are detached from their supporting cells, at which point they undergo apoptosis. Angiopoietin 2 (Ang-2) expression is induced in the co-opted vessels prior to their regression. Ang-2 is involved in physiologic vessel remodeling in a VEGF-dependent manner (see succeeding discussion). Co-option of existing vessels may be an important method for obtaining a vascular supply in early tumors. Its extent is controlled by the local production of VEGF, Ang-1, and Ang-2.
Intussusceptive microvascular growth, the longitudinal separation of existing vessels into daughter vessels, is another mechanism of enhancing blood supply to tumors. In this way, the network’s complexity and efficiency improves, with no need for endothelial cell proliferation.
Vasculogenic mimicry, which has been observed mostly in melanomas, is the ability of cancer cells to transform into endothelial-like cells in specific sites, thus forming blood vessels made of cancer cells.
38 The importance of these alternative mechanisms of vascular supply in cancer growth is not known.
Role of Immune System Cells in Angiogenesis The stroma of cancer is infiltrated by immune system cells in varying proportions. The role of the immune system in the progression of cancer is not obvious but seems to be context dependent. The immune system has a cancer-inhibitory effect, as evidenced by the high risk of cancer in immunocompromised patients. In addition, a correlation was found between increased effector memory T-cell infiltration of the tumor and a good prognosis in colon cancer patients.
70 However, in many other settings, the immune system apparently contributes to cancer progression. The adaptive immune system can mount an antitumor response in some conditions. In contrast, innate immune system cells may be more commonly recruited by cancer cells and function in a pro-cancer manner.
71 Neutrophilic infiltration of tumors is common, but its clinical significance is not clear. In bronchoalveolar carcinoma, it has been associated with a poor prognosis,
72 but data on its importance in other cancers are limited. Lung cancer-infiltrating lymphocytes exert an anticancerous effect, as in colon cancer. High densities of CD4
+ and CD8
+ lymphocytes in the stroma of NSCLC tumors have been found to be associated with a good prognosis.
73,
74
Monocytes circulate in the blood; once recruited to sites of tissue inflammation, they differentiate into macrophages. Various chemoattractants play a role in the chemotaxis of monocytes into tumors, possibly mostly to hypoxic areas of tumors.
75 Macrophages constitute a major subset of the immune system cells that populate the tumor stroma: tumor-associated macrophages (TAMs). TAMs seem to promote the progression of cancer.
76 Unlike classically activated macrophages (M1 macrophages), TAMs have a poor antigen-presenting ability and produce factors that suppress T-cell proliferation and activity. The chemokines and chemokine receptors profile they express is adapted for scavenging for debris, promoting cell migration and angiogenesis, and repairing and remodeling wounded or damaged tissues. Cancer microenvironment exposure to interleukin (IL)-4 and IL-10 can induce monocytes to develop into TAMs (otherwise known as polarized type II [alternatively activated] or M2 macrophages).
77 Proangiogenic monocytes that localize in tumors are also characterized as Tie-2 expressors.
46 Macrophage infiltration was found to be associated with vessel density in several types of cancer. In a mouse model of breast cancer, depletion of macrophages inhibited the angiogenic switch and cancer progression. Increased macrophage infiltration was correlated with earlier tumor progression.
78IL-1
β is a proangiogenic cytokine that depends on macrophage recruitment to tumor sites for its angiogenic effect.
79 MMP9 release by macrophages, leading to mobilization of VEGF,
80 is a mechanism in which the infiltration of macrophages or other myelomonocytes
81 induces angiogenesis. In addition, IL-8/CXCL8, another proangiogenic factor, was upregulated in both cancer cells and macrophages when these two cell types were cultured together, and IL-8/CXCL8 mRNA levels in lung cancer specimens were correlated with MVD and poor patient prognosis.
82 Relating an angiogenic response to inflammation, which is common in cancer, IL-1
α was shown to recruit VEGF-expressing inflammatory cells. VEGFR-2 blockage prevented this angiogenic response.
83 Regardless of the available data on the contribution of macrophages to angiogenesis, tumor islet infiltration by macrophages was a good prognostic factor in a study of 175 NSCLC patients. On the other hand, stromal macrophage infiltration was associated with a poor prognosis.
84 These results were reproduced in a study of 199 NSCLC patients.
85 Importantly, the studies that showed a positive prognostic effect of macrophages in lung tumors differed from earlier studies by differentiating between stromal and tumoral macrophages.
Natural killer (NK) cells are another part of the innate immune system that have important interactions with cancer and cancer-induced angiogenesis. NK cells recognize and lyse cancer cells and are thought to have important roles in immune surveillance against cancer. IL-12 is an antiangiogenic agent that depends on NK cell recruitment to affect cancer angiogenesis.
86 NK cells secrete interferon-
γ, causing inhibition of endothelial cell proliferation; this is probably the major mechanism of NK cells’ antiangiogenic effects.
87Mast cells were found to be essential for tumor progression in a mouse model of squamous cell carcinoma and were required for neoangiogenesis.
88 Mast cell infiltration was correlated with MVD in a study of NSCLC specimens.
89 However, mast cell tumor infiltration was associated with a good prognosis in NSCLC specimens.
84 More detailed studies of the role of mast cells in lung cancer are required. In light of the inhibitory effect of most anticancer treatments on the immune system, further insight is required into the effect of immune system cells on angiogenesis and cancer progression.
Tumor Stroma-Dependent Effects The stroma of tumors is more than a mechanical scaffold; stromal cells seem to be reprogrammed by cancer cells to participate in cancer progression. A mouse model demonstrated activation of the VEGF gene promoter in tumor stroma fibroblasts.
90 CAFs also contribute to cancer progression,
91 apparently by activating angiogenesis. Activation of hepatocyte growth factor (HGF)-c-Met signaling is another role of the stroma in tumor angiogenesis (see succeeding discussion).
92
MMPs are a family of Zn2++ proteases that are produced mostly by stromal fibroblasts and by cancer and endothelial cells. These proteases have important roles in angiogenesis. MMP2 and MMP9, for example, mediate the breakdown of collagen type IV, a major component of the vascular basement membrane. The mobilization of growth factors, including
VEGF
80 and additional angiogenic molecules from the ECM, is another angiogenic activity of MMP. This mobilization may be essential in the initial stages of cancer, becoming less important as the tumor progresses and alternative sources of VEGF become available. MMP9 was also shown to be required for the recruitment of bone marrow-derived cells into the tumor microenvironment, for the maturation of tumor vasculature, and for pericyte coverage.
93 In contrast, at later stages of tumor growth and MMP activity, the dominant end products of MMP protein cleavage are antiangiogenic factors.
94 Although MMPs are correlated with angiogenic activity in lung cancer, general MMPs inhibition did not improve the clinical outcome of NSCLC patients.
95 Modification of specific MMP(s) might be required for impacting clinical end points.
Vascular Basement Membrane The ECM that envelops endothelial cells, and within which pericytes are embedded, is called the vascular basement membrane. The major collagen that constitutes the basement membrane is collagen IV, which has the unique ability to self-assemble into sheets. Additional components are laminins, which bind cell membrane anchors such as integrins on one side and ECM collagen on the other side. The mature basement membrane signals differentiation and reduced proliferation to adjacent endothelial cells. The same protein constituents, while being deposited as a new basement membrane, present different molecular moieties to the cells around them. Through integrins, they provide proliferation and migration signals. The ECM structural components also include molecular messengers, such as endostatin, that can be proteolytically released from collagen XVIII and functions as an antiangiogenic effector. Arrestin, canstatin, and tumstatin are also collagen-derived antiangiogenic molecules. On the other hand, triple-helix fragments of collagen IV activate endothelial cell migration. Therefore, specific molecules in the ECM/basement membrane can convey different messages at different phases of tumor growth. Deposited by the resident cells, the ECM/basement membrane is an important manner of cell-cell indirect communication. This adds another level of complexity to the cellular events occurring in the process of new blood vessel formation.