(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Mitochondria are not generated de novo but grow and divide adding newly synthesized constituents to preexisting mitochondrial compartments. Mitochondria have their own compact circular genome and multiprotein machineries that mediate replication, transcription, and translation. During the past two decades, the core components implicated in these processes have been identified, and several molecular mechanisms underlying mtDNA dynamics have been proposed. The ability to repair certain types of mtDNA lesions using BER, mismatch repair, and recombinational repair pathways has been also demonstrated, and several proteins implicated in these pathways have been identified.
The vast majority of mitochondrial proteins, including components of the OXPHOS machinery, is encoded by the nuclear genome, synthesized on cytosolic ribosomes, and imported through complex pathways. At least four major pathways are exploited for biogenesis of mitochondrial proteins, including the presequence pathway, the carrier protein pathway, the redox-regulated import pathway, and the β-barrel protein pathway. Biogenesis of the mitochondrial membranes relies on the highly coordinated synthesis, import, and assembly of proteins and phospholipids. However, in contrast to the biogenesis of mitochondrial proteins, the mechanisms of mitochondrial phospholipid biosynthesis and trafficking are much less characterized.
The coordinated expression of a subset of genes in the nucleus and mitochondria is orchestrated by the intricate network of transcription factors and cofactors. While nuclear-encoded mitochondrial factors TFAM, TFB1M, TFB2M, and mTERF govern mtDNA maintenance, respiratory factors NRF1 and NRF2, ERRs, PPARs, and PGC-1 transcriptional coactivators regulate expression of multiple OXPHOS subunits, FA transporter, and FA oxidation enzymes. The PGC-1 family members, PGC-1α, PGC-1β, and PRC, play a central role in integrating various signals and coordinating a variety of transcription factors to orchestrate mitochondrial biogenesis and respiratory function. A central role of the PGC-1 pathway in orchestrating energy metabolism in energy-demanding myocardium and the recent finding that it is downregulated in the failing heart suggest that this regulatory circuit could be a potential target for treatment of myocardial disorders. At present, many fundamental questions still remain unanswered regarding the molecular mechanisms of mitochondrial biogenesis to therapeutically target this process in the treatment of cardiovascular diseases.
Introduction
Mitochondria are unusual organelles as they are not generated de novo but grow and divide adding newly synthesized constituents to preexisting mitochondrial compartments. Mitochondria have their own compact genome (mtDNA) and multiprotein assemblies involved in replication, transcription, and translation. However, the vast majority (more than 98%) of mitochondrial proteins, including components of the OXPHOS machinery, is encoded by the nuclear genome (nDNA), synthesized on cytosolic ribosomes, and must be imported through complex pathways (Fig. 4.1) [1, 2].
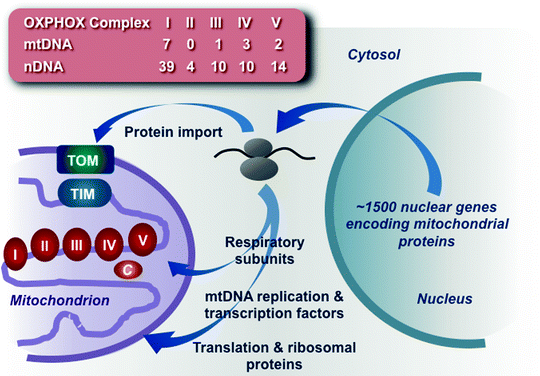
Fig. 4.1
Schematic summary of mitochondrial biogenesis. The vast majority of mitochondrial proteins, including components of the oxidative phosphorylation (OXPHOS) machinery, mtDNA replication and transcription factors, and mitochondrial ribosomal and translation proteins, is encoded by the nuclear genome, synthesized on cytosolic ribosomes, and imported through complex pathways. The number of protein subunits encoded by mitochondrial (mtDNA) and nuclear (nDNA) genomes is shown for each of the five OXPHOS complexes. c cytochrome c, TIM translocase of the mitochondrial inner membrane, TOM translocase of the mitochondrial outer membrane
The number of mitochondria per cardiomyocyte may vary significantly, depending on the metabolic requirements. Complex processes of mitochondrial fusion and fission contribute to mitochondrial biogenesis. Mitochondrial fusion contributes to the even distribution of mitochondrial constituents, whereas fission increases the number of organelles and targets damaged organelles to mitophagy [3–6]. Moreover, the dynamic interconnected mitochondrial networks produced by fusion are characteristic for energy-demanding cardiomyocytes. Dysfunctional mitochondria are selectively eliminated by the specific autophagic pathway, mitophagy [6]. Finely tuned balance between mitochondrial biogenesis and mitophagy is critical to maintain mitochondrial number as well as for quality control [7–9].
Emerging evidence suggests that mitochondrial biogenesis is finely tuned largely at the level of transcription. The coordinated expression of a subset of genes in the nucleus and mitochondria is governed by an intricate network of transcription factors and cofactors [10–14]. The master regulator of mitochondrial biogenesis is the peroxisome-proliferator-activated receptor γ coactivator-1α (PGC-1α). PGC-1α induces transcriptional activity of the nuclear respiratory factors NRF1 and NRF2. Activated NRF1 and NFR2 in turn regulate expression of multiple components of the respiratory complexes I–V, while expression of the fatty acid (FA) transporter and FA oxidation (FAO) enzymes is controlled by the peroxisome-proliferator-activated receptors (PPARs) [12, 15–17]. The estrogen-related receptors (ERRs) also contribute to the transcriptional regulation of ATP synthesis and the transport across the mitochondrial membranes [18]. In addition, nuclear-encoded transcription factors TFAM, TFB1M, and TFB2M; RNA polymerase; and a termination factor mTERF control replication, transcription, and maintenance of mtDNA [19].
Thus, mitochondrial biogenesis is an elaborate, finely tuned process, which requires intensive mitochondrial-nuclear communication to coordinate expression and assembly of numerous proteins encoded by the two genomes. While the crucial role of mitochondria in myocardial function has long been appreciated, the molecular mechanisms of regulation of mitochondrial biogenesis in the myocardium are largely unknown. Multiple genetic and environmental factors, alterations in metabolic signaling and oxidative stress, aging, and hyperglycemia can affect cardiac mitochondrial biogenesis contributing to the pathogenesis of various cardiovascular diseases (CVD).
Mitochondrial Genome: Structure and Dynamics
Mitochondrial DNA
Human mitochondrial DNA (mtDNA) is a 16,569-bp circular negatively supercoiled double-stranded molecule, formed by a light (L-; pyrimidine-rich) and a heavy (H-; purine-rich) strand. The mtDNA strands have slightly different densities, due to their different base composition, and can be separated by alkaline CsCl gradient centrifugation [20]. In mammalian cells, mtDNA is present in approximately 1,000 copies per cell and comprises only 0.1–2% of the total DNA. mtDNA encodes 37 transcripts: 13 polypeptides, 2 ribosomal RNAs (rRNAs), and 22 transfer RNAs (tRNAs). Seven mtDNA-encoded proteins represent subunits (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6) of Complex I, one (cytochrome b) is a subunit of Complex III, three are subunits (CO1, CO2, and CO3) of Complex IV, and two are subunits (ATP6 and ATP8) of Complex V (Fig. 4.2) [21, 22]. The rest of the components of the respiratory complexes and approximately 1,500 other mitochondrial proteins are encoded by the nuclear genome and synthesized in the cytosol.
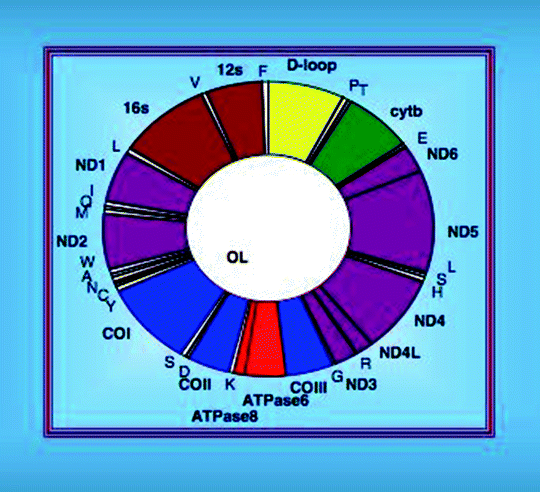
Fig. 4.2
Organization of the human mitochondrial DNA
The mitochondrial genome is highly compact with most protein and rRNA genes being separated by tRNA genes. The mtDNA transcription occurs in both directions with the open reading frames on either strand. Most of genetic information is encoded in the H-strand, while the L-strand encodes only one polypeptide and eight tRNAs. A noncoding region (NCR) of approximately 1 kb long is located between the tRNAPro and tRNAPhe genes and contains the L-strand promoter (LSP), H-strand promoter (HSP), origins of replications (OH, Ori-b, and Ori-bL), evolutionary conserved sequence blocks (CSB), and a replication-termination-associated sequence (TAS). A 650-nucleotide single-stranded DNA (7S DNA), generated by replication initiating at OH and terminated at TAS, is annealed to the parental L-strand displacing the parental H-strand to form a triple-stranded DNA structure, D-loop.
There are several additional unique characteristics of the mitochondrial genome. Mitochondrial genes contain no introns, and intergenic sequences are absent or limited to a few bases. Some genes overlap and have in some instances no termination codons [23]. The mitochondrial genetic code differs from the universal genetic code. In mitochondria, AUA encodes Met instead of Ile, and the universal termination codon UGA encodes Trp, while AGA and AGG, which normally encode Arg, are recognized as termination codons. Moreover, there is a higher flexibility in the pairing of the third base of mitochondrial anticodons compared to their cytosolic counterparts. This allows the limited set of 22 tRNA species encoded by human mtDNA to be sufficient to translate all mitochondrial polypeptides. Finally, mtDNA is almost exclusively maternally inherited exhibiting non-Mendelian inheritance pattern, since the mitochondrial phenotype is determined by multiple mtDNA copies present in the mitochondrion [22, 24, 25].
Mitochondrial Nucleoid
Since mitochondria do not have histones and mtDNA displays relatively high mutation rates, mtDNA has initially been presumed to be “naked.” However, extensive genetic, biochemical, and cell biological studies first in yeast and later in mammals, including humans, have demonstrated that mtDNA is packaged into DNA–protein complexes termed by analogy to those in bacteria, nucleoids. According to the current paradigm, (a) each cell type, depending on growth conditions and developmental/differentiation stage, contains multiple nucleoids (tens in yeast to hundreds in mammals), each containing several copies (up to ten in human cells) of mtDNA and various nucleoid-associated proteins, and (b) each nucleoid represents a relatively stable genetic element [26–29]. Finally, while mitochondrial nucleoids are located within the mitochondrial matrix, they appear to be tethered to the MIM [30–32].
Multiple DNA-binding proteins have been demonstrated to be specifically associated with mtDNA in nucleoids (Table 4.1). The transcription factor A of mitochondria (TFAM, also known as mtTFA; the mammalian homolog of yeast Abf2p) and the mitochondrial single-stranded DNA-binding protein (mtSSB; the mammalian Rim1p homolog) are the most abundant and tightly nucleoid-associated proteins [33–36].
Table 4.1
Human nucleoid-associated proteins
Human protein | Function |
|---|---|
Core nucleoid-associated proteins | |
TFAM | mtDNA dynamics and maintenance |
mtSSB | |
POLγA and POLγB | mtDNA dynamics and maintenance |
POLRMT | |
TFB2M | |
Twinkle helicase | |
DHX30 helicase | |
SUPV3L helicase | |
HMG protein A1 | |
BRCA1 | |
Leucine-rich protein 130 | |
Polymerase δ-interacting protein 38 | |
HSP60 | Molecular chaperones |
HSP70 | |
HSP90-β | |
Lon protease | Proteases and metabolic enzymes |
CLPX protease | |
Prohibitins 1 and 2 | |
Aspartate aminotransferase | |
Adenosine nucleotide transferase | |
ATPase subunits β and O | |
Carbamoyl phosphate synthetase 1 | |
Carnitine palmitoyltransferase 1A | |
Serine hydroxymethyltransferase | |
2,4-dienoyl-CoA reductase | |
Hydroxyacyl dehydrogenase A | |
Malate dehydrogenase | |
NADH dehydrogenase subunits S1, 1a9, and 30K | |
Peroxide reductase 3 | |
Actin | Cytoskeletal components |
Vimentin | |
ATAD3 | Various functions |
Bcl-XL-binding protein v68 | P53 |
Squamous cell carcinoma antigens 1 and 2 | |
TFAM is an abundant high-mobility group (HMG) protein, originally identified as a mitochondrial transcription factor. It acts as a homodimer that binds mtDNA at an average of one TFAM dimer per 35–40 bp of mtDNA [37–40]. The presence of TFAM in nucleoids in human cells has been confirmed by immunofluorescence approach [26, 38, 41]. It plays the architectural role in mtDNA packaging similar to the role of nuclear histones in chromatin organization. TFAM is also involved in mitochondrial transcription and regulation of mtDNA copy number.
mtSSB has been shown to colocalize with mtDNA in situ and copurify with nucleoids isolated from human cells [26, 42–44]. Drosophila mtSSB is required for mtDNA replication and maintenance [45]. It has been suggested that mtSSB plays a role in stabilization of the D-loop structure and replication intermediates.
Analysis of isolated preparations of mitochondrial nucleoids has identified several additional nucleoid-associated proteins, including the mtDNA polymerase γ catalytic and accessory subunits, DNA helicase Twinkle, and mitochondrial RNA polymerase [26]. Moreover, it has recently been reported that a subfraction of a D-loop-binding protein ATAD3 is associated with mtDNA and plays a role in the organization of multiple mtDNA copies and segregation [46, 47]. Given all these proteins appear to be involved in mtDNA maintenance, their association with mitochondrial nucleoid is not unexpected. However, surprisingly in isolated nucleoid preparations, several proteins located in the MIM have been found, including ANT and components of the respiratory Complexes I and V [43, 44]. While it is tempting to speculate that these MIM proteins anchor mtDNA to the MIM, it is unclear whether they play this role or are rather nonspecific contaminants. Lastly, several signaling proteins, such as prohibitins 1 and 2 and p53 implicated in cell cycle control, apoptosis, signal transduction, repair, and lifespan regulation, represent other potential nucleoid components [43, 44, 48–50].
Thus, mtDNA is packaged in mitochondrial nucleoids, which represent the molecular unit of mitochondrial genetics. Multiple proteins contribute to this mtDNA packaging; however, the precise function of many nucleoid-associated proteins is not clear. Core nucleoid proteins, such as TFAM and mtSSB, are always associated with most mtDNA, while many other nucleoid-associated proteins can be only transiently associated with a subset of nucleoids, depending on the specific cellular conditions.
Replication of Mitochondrial DNA
Mitochondrial Replisome
Mitochondria have a multiprotein machinery, mitochondrial replisome, capable to promote mtDNA replication. All components of mitochondrial replisome in mammals are encoded by nuclear genes, translated on cytosolic ribosomes, and imported into mitochondria. Currently, only major players involved in mtDNA replication are known. They include the DNA polymerase γ holoenzyme, mtSSB, the replicative DNA helicase Twinkle, and the mitochondrial RNA polymerase (POLRMT) (Fig. 4.3) [51–55]. In vitro reconstitution experiments have demonstrated that these replicative proteins represent a minimal mitochondrial replisome capable of replicating the full length of the mtDNA molecule at a rate not significantly different than that found in vivo [56, 57].
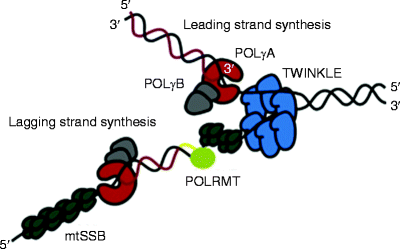
Fig. 4.3
Schematic representation of the mitochondrial replisome. The Twinkle helicase (shown in blue) slides in a 5′ to 3′ direction unwinding double-stranded DNA (dsDNA). The mitochondrial single-stranded DNA-binding protein (mtSSB, dark green) stabilizes exposed single-stranded mitochondrial DNA (mtDNA) regions and stimulates mtDNA synthesis by the DNA polymerase γ holoenzyme [POLγA (red) and POLγB (gray)]. The mitochondrial RNA polymerase (POLRMT, light green) synthesizes the RNA primer (yellow line) required for lagging-strand DNA synthesis (From Wanrooij and Falkenberg [83] with permission from Elsevier)
DNA Polymerase γ
The only DNA polymerase found in mitochondria is the DNA polymerase γ (POLγ) [58]. The POLγ holoenzyme is a 245-kDa heterotrimer composed of one catalytic subunit (POLγA also referred to as POLG1) and two accessory subunits (POLγB also called POLG2) [59, 60]. POLγA has three polymerase motifs mediating DNA synthesis, and a 3′ → 5′-exonuclease domain promoting proofreading during replication. POLγB functions as a processivity factor (i.e., it enhances the ability of an enzyme to repeatedly continue its catalytic function without uncoupling from its substrate); one POLγB subunit located close to POLγA enhances POLγA–DNA binding, while the more distantly located POLγB subunit accelerates polymerization rate [61–64]. POLγA levels and activity do not change significantly during development and aging [65]. Moreover, unlike TFAM, the DNA polymerase γ is stably expressed in the absence of mtDNA [66].
Mitochondrial Single-Stranded DNA-Binding Protein
Human mtSSB is homologous to the Escherichia coli SSB, and close homologs have been identified in yeast, Drosophila, Xenopus laevis, and mouse [53, 67–70]. mtSSB stabilizes exposed single-stranded mtDNA regions and stimulates primer recognition and processivity of POLγ at the replicative fork, facilitating thereby mtDNA synthesis [68, 71, 72]. Moreover, human mtSSB stimulates Twinkle helicase activity [73]. New evidence suggests direct interactions between mtSSB and other mitochondrial replicative proteins at the replication fork. Lastly, its levels correlate with mtDNA content and are upregulated during early postnatal development of the heart [65].
Twinkle
The acronym Twinkle stands for T7 gp4-like protein with intramitochondrial nucleoid localization. The C-terminal region of Twinkle is homologous to the T7 phage primase/helicase protein gp4 essential for T7 DNA replication [54]. Twinkle exhibits a 5′ → 3′ DNA helicase activity, which is essential in vivo [73–75]. While Twinkle has homology to T7 gp4, human Twinkle lacks critical amino acids observed in related primases and does not have primase activity [54, 76]. Twinkle can form hexa- and heptamers; its linker region, connecting the N- and C-terminal regions, is responsible for this ability [77, 78].
In contrast to the gp4 protein and other replicative helicases, Twinkle prefers a dsDNA substrate with a 5′-loading site and a short 3′-tail to initiate DNA unwinding [73, 74, 79, 80]. However, the functional significance of dsDNA preference of Twinkle is unclear. Intriguingly, the Twinkle helicase motifs have similar organization as a main E. coli recombinase, RecA protein, and a role for Twinkle in recombination has been suggested. Consistent with this hypothesis, its overexpression has led to significant decrease in mtDNA recombination intermediates in human cardiomyocytes [81, 82].
Currently, 31 different mutations in the TWINKLE gene have been identified, resulting in depletion of mtDNA leading to mitochondrial dysfunction (Fig. 4.4) [83]. These TWINKLE mutations are associated with neuromuscular abnormalities. All dominant TWINKLE mutations are responsible for autosomal dominant progressive external ophthalmoplegia (adPEO) characterized by exercise intolerance and muscle weakness, and the ocular muscles are most severely affected [54, 84]. AdPEO patients accumulate deletions in mtDNA in postmitotic tissues [54].
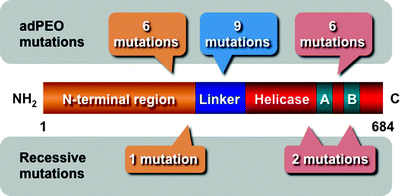
Fig. 4.4
Scheme of the molecular structure of the Twinkle helicase and disease-associated mutations. The N-terminal region, linker, and helicase domain with Walker A (A) and B (B) motifs are depicted. Dominant mutations responsible for autosomal dominant progressive external ophthalmoplegia (adPEO) and recessive mutations are also schematically shown
Mitochondrial RNA Polymerase
Mitochondrial RNA Polymerase (POLRMT) responsible for mitochondrial transcription is a unique RNA polymerase distinct from the nuclear counterparts and structurally similar to the T7 phage RNA polymerase [55]. POLRMT is not only essential for transcription, but it also synthesizes the RNA primers to prime leading-strand mtDNA synthesis at the origin of heavy-strand replication (OH) [57, 85, 86]. Transcription from the light-strand promoters (LSP) synthesizes genome length transcripts, which subsequently are processed to yield the individual mRNA and several tRNA species [23, 87]. In mammalian cells, H-strand DNA replication initiation events usually are terminated approximately 600 bp downstream of OH. The terminated strand remains stably annealed to the parental DNA molecule generating a triplex structure, D-loop, with a displaced parental H-strand [88, 89].
The role of POLRMT as the leading-strand primase responsible for primer synthesis at OH has been well established. However, the enzyme responsible for initiation of lagging-strand mtDNA synthesis has remained elusive. Emerging evidence suggests that POLRMT can also prime DNA synthesis at the origin of the light-strand replication (OL) acting as a lagging-strand primase. According to the classical model, upon passage of the leading-strand replication machinery, single-stranded OL is exposed and forms a stem-loop structure [90, 91]. mtSSB cannot bind to the stem-loop, while POLRMT can initiate RNA primer synthesis from a poly-dT repeat of the OL stem-loop. After approximately 25-nt primer synthesis is completed, POLRMT is replaced by POLγ to initiate lagging-strand DNA synthesis. Importantly, this process can be reconstituted in vitro, and POLRMT depletion in human cells has resulted in delay in lagging-strand synthesis [57, 92].
Finally, a human ligase III gene has been reported to encode both nuclear and mitochondrial isoforms [93]. Although a role for the ligase III in mtDNA repair has been suggested, its role in mtDNA replication remains to be determined.
mtDNA Replication Mechanisms
A few models of mitochondrial replication in mammals have been proposed, although the precise molecular mechanisms of mtDNA replication are a matter of open debate [83, 94].
According to the strand-displacement model (SDM), first proposed in 1972, leading and lagging strands are synthesized asymmetrically and unidirectionally from separate origins, OH and OL, respectively [95–98]. The mitochondrial replication fork initiates the synthesis from a POLRMT-synthesized primer and often arrests at the termination-associated sequence (TAS) generating a D-loop of approximately 650 bp in human cells [88, 89, 98]. When the leading-strand synthesis continues unidirectionally beyond the TAS, it leaves behind a single-stranded region of the parental H-strand. mtSSB binds to this ssDNA preventing nonspecific POLRMT-mediated lagging-strand priming. After traversing approximately 2/3 of the genome, the replication fork exposes the lagging-strand replication initiation site, OL. Displaced parental H-strand adopts a stem-loop structure, and POLRMT initiates RNA primer synthesis from the poly-dT stretch of this region. POLγ replaces POLRMT and initiates the lagging-strand mtDNA synthesis (Fig. 4.5a).
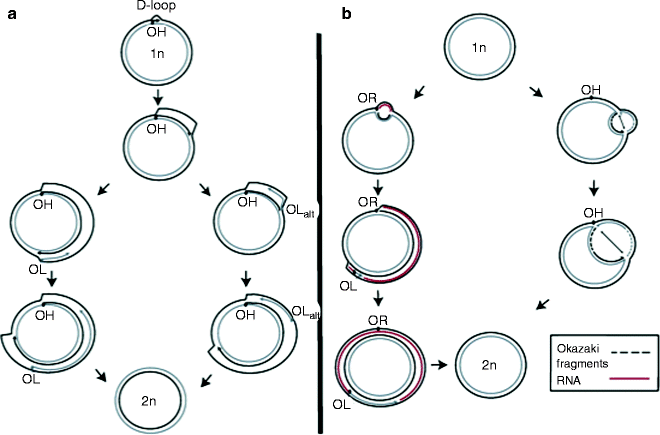
Fig. 4.5
Main models for mammalian mitochondrial DNA replication. (a) Strand-displacement replication model; (b) ribonucleotide incorporation throughout the lagging strand (RITOLS) model (left pathway) and the strand-coupled model (right pathway). See text for details (From Wanrooij and Falkenberg [83] with permission from Elsevier)
Based on the finding of double-stranded replication intermediates generated by coupled leading- and lagging-strand synthesis, which were RNase H sensitive, two additional mtDNA replication models have been proposed [99–102]. In the ribonucleotide incorporation throughout the lagging strand (RITOLS) model, the leading strand (H-strand) is replicated unidirectionally from a discrete origin. The lagging-strand synthesis occurs with delay, like in the SDM; however, ssDNA covered by RNA instead of mtSSB generating theta-type, double-stranded RITOLS intermediates sensitive to RNase H (Fig. 4.5b). This RNA may be processed to generate multiple short RNA primers, which can be used to initiate discontinuous lagging-strand replication [102].
According to the strand-coupled model, in contrast to the RITOLS, mtDNA intermediates are fully dsDNA similar to nDNA replication intermediates; hence, this mechanism is also termed as conventional, strand-coupled Okazaki fragment associated (COSCOFA) replication [102]. In the COSCOFA, both leading- (H-strand) and lagging-strand (L-strand) syntheses initiate bidirectionally from multiple origins at a broad initiation zone (Ori-Z) downstream of OH [103]. The lagging strand is synthesized without delay via most likely Okazaki fragment-associating replication. Both replication forks proceed till they arrest at OH (Fig. 4.5b). Although the existence of Okazaki fragments during mtDNA replication has not yet been shown, mammalian mitochondria possess enzymatic machinery for their processing [104]. The role of mtSSB in both RITOLS and COSCOFA replications remains to be determined. Different mammalian tissues may exploit different mtDNA replication mechanisms: COSCOFA intermediates have predominantly been found in the heart, skeletal muscle, and in cells amplifying mtDNA after its depletion, while RITOLS intermediates have been found in liver, kidney, and cultured cells [78, 82, 102, 105].
Transcription of Mitochondrial DNA
The identification and characterization of trans-acting factors involved in mtDNA replication, transcription, and translation have generally been hampered by their extremely low concentrations as well as by difficulties to obtain highly purified mitochondrial preparations devoid of nuclear/cytosolic contaminants. However, major central players have recently been identified, and their functions in mtDNA maintenance have been characterized first in yeast and subsequently in mammalian including human cells.
Mitochondrial Transcription Machinery
The core human mitochondrial transcription machinery consists of mitochondrial RNA polymerase (POLRMT), an HMG-box transcription factor TFAM, and two mitochondrial transcription factors B1 and B2 (TFB1M and TFB2M, also known as mtTFB1 and mtTFB2, respectively) (Fig. 4.6) [106].
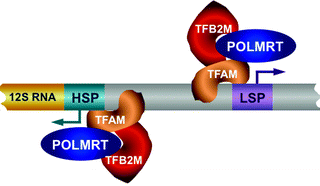
Fig. 4.6
The core human mitochondrial transcription machinery. Mitochondrial RNA polymerase (POLRMT), high-mobility group (HMG) box transcription factor A of mitochondria (TFAM), and transcription factor B2, mitochondrial (TFB2M) are depicted. Mitochondrial DNA (mtDNA) transcription is initiated by the TFAM binding upstream of the light-strand promoter (LSP) and heavy-strand promoter (HSP). TFAM bend and unwind mtDNA allowing the loading of TFB2M and POLRMT on the promoter to mediate mtDNA transcription bidirectionally
POLRMT
The human POLRMT gene cloned in 1997 encodes a 140-kDa single-subunit protein, containing two large regions characteristic to eukaryotic mtRNA polymerases [55]. The N-terminal part of the protein contains a 41-amino acid targeting sequence and two putative pentatricopeptide repeats (PPR) found in proteins involved in RNA processing in mitochondria and chloroplasts [107, 108]. An alternatively spliced POLRMT isoform missing the N-terminal 262 amino acids, including mitochondrial targeting signal, is located in the nucleus and represents the nuclear RNA polymerase IV; however, its function is not known [109].
The C-terminal portion of POLRMT contains several conserved sequence motifs essential for its catalytic activity and is homologous to the bacteriophage T3/T7 RNA polymerase [110]. However, unlike the bacteriophage T7 RNA polymerase, POLRMT is uncapable to initiate transcription on its own and requires assistance of TFAM and TFB1M/TFB2M [106, 111].
TFAM
TFAM is a multifunctional protein involved in mtDNA packaging, replication, and transcription [38, 39, 112]. Human TFAM was the first mitochondrial transcription factor identified in mammalian cells [113]. TFAM is a member of the high-mobility group (HMG) family of proteins, which are able to bend, wrap, and unwind DNA [114, 115]. TFAM contains two HMG-box DNA-binding domains separated by a linker region and a short C-terminal tail, which is required for specific binding to the promoter region and for interaction with TFB1M/TFB2M and activation of transcription [116, 117]. Like other HMG proteins, TFAM can bind, bend, and unwind mtDNA in a sequence-nonspecific way; for nonspecific binding, its C-terminal part is not essential. However, mtDNA transcription is initiated by the sequence-specific binding of TFAM dimer upstream of the HSP and LSP [118, 119].
TFB1M and TFB2M
TFB1M and TFB2M have originally been identified as homologs of single yeast mitochondrial transcription factor mtTFB, essential for mtDNA transcription in this organism [111]. Like their yeast homolog, TFB1M and TFB2M can assemble into a complex with POLRMT and stimulate transcription in the presence of TFAM in vitro [106]. They also display sequence homology to the bacterial rRNA dimethyltransferases, which promote dimethylation of two adjacent adenine residues at the 3′ end of rRNA during ribosome biogenesis [106, 111]. Consistently, human TFB1M is capable to mediate this reaction in vitro [120]. rRNA dimethyltransferase activity of TFB2M is less efficient than that of TFB1M [121]. Mutations in conserved methyltransferase motifs that affect TFB1M/TFB2M rRNA dimethyltransferase activity do not change their ability to stimulate mtDNA transcription, suggesting that these functions are independent [117].
In the presence of TFB2M, TFB1M appears to be not essential for mtDNA transcription; however, its loss in mice results in embryonic lethality. Moreover, TFB1M defect in the heart has led to mitochondrial deficiency caused mainly by impaired mitochondrial translation [122]. Loss of TFB1M dimethyltransferase activity appears to be responsible for this defect, since dimethylation of 12S rRNA is essential for assembly of the ribosomal subunits [123]. Accordingly, TFB1M silencing has led to significant decrease in 12S rRNA levels, while its overexpression has resulted in 12S rRNA hypermethylation associated with impaired mitochondrial biogenesis and cell death [124].
In contrast, TFB2M displays significantly greater transcriptional stimulatory activity than TFB1M and plays predominant role in mtDNA transcription [106]. Consistently, overexpression of TFB2M but not TFB1M has led to elevated steady-state levels of mtDNA transcripts, while disruption of its rRNA dimethyltransferase domain has had no apparent effects [124, 125]. Thus, emerging evidence suggests that these proteins have diverged but partially overlapping functions: TFB1M predominantly regulates mitochondrial translation, while TFB2M predominantly stimulates mitochondrial transcription.
Regulation of Mitochondrial Transcription
In human cells, the noncoding region (NCR), also known as D-loop (spanning nucleotides 16,024–16,576), contains the control elements for both mtDNA transcription and replication. Each strand contains the light-strand promoter (LSP), two heavy-strand promoters (HSP1 and HSP2), and the origin of replication for the heavy strand (OH) (Fig. 4.7) [126, 127]. mtDNA transcription from the LSP and HSP2 generates long polycistronic precursor RNAs, which are subsequently processed by RNase P to produce mature mRNAs, rRNAs, and tRNAs [23, 87, 128, 129]. Transcription from the HSP1 generates a truncated transcript, which encodes 12S and 16S rRNA and two tRNAs [130]. Finally, as it has already been discussed, mtDNA transcription initiated by POLRMT from the LSP generates a primer for mtDNA replication, coupling thereby mitochondrial gene expression and mtDNA maintenance [131].
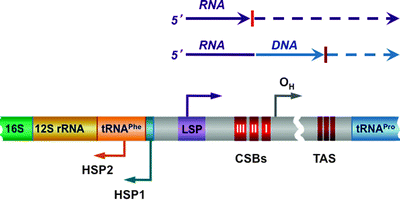
Fig. 4.7
Scheme of the D-loop regulatory region. The conserved sequence blocks (CSB I, II, and III) are located downstream of the light-strand promoter (LSP), while the conserved termination-associated sequence (TAS) elements are located at the 3′ of the nascent D-loop strands. Two heavy-strand promoters (HSP1 and HSP2) and the origin of replication for the heavy strand (OH) are also shown. Mitochondrial DNA (mtDNA) transcription from the LSP and HSP2 generates long polycistronic precursor RNAs. Transcription from the LSP generates also RNA primer for mtDNA replication; transition from the primer to the newly synthesized DNA has been mapped within or near CSB II. See text for details
Transcription initiation is followed by the elongation step. The mitochondrial transcription elongation factor (TEFM) has recently been identified to interact with POLRMT enhancing its processivity. Consistent with an essential role of TEFM in mitochondrial transcription, its deficiency has led to reduced mtDNA transcript levels [132].
Promoter Recognition and Transcription Initiation
It is generally believed that the simultaneous presence of TFAM and the POLRMT–TFB2M heterodimer is required for promoter recognition and initiation of transcription. TFAM homodimer binds specifically to a region upstream of the HSP and LSP [119]. The C-terminal tail of TFAM, required for specific binding to the promoter region, also mediates direct protein–protein interaction with TFB2M, recruiting thereby POLRMT to the transcription initiation sites [57, 117]. In addition, TFAM due to its HMG-box DNA-binding domains mediates bending and unwinding of mtDNA helix. These structural DNA alterations expose the promoter region and may facilitate its recognition and initiation of transcription by the POLRMT–TFB2M complex.
Thus, the core machinery needed for mitochondrial transcription initiation is traditionally regarded as a three-component system composed of POLRMT, TFB2M, and TFAM [131, 133]. However, more recently, it has been demonstrated using a human recombinant mitochondrial transcription system that POLRMT and TFB2M can initiate transcription from the HSP1 and LSP even in the absence of TFAM, albeit with significantly lower efficiency [134]. Importantly, in vitro TFAM-independent transcription from the HSP1 has been equal or higher than from the LSP, while TFAM-dependent transcription has been higher from the LSP than from the HSP1. Moreover, additional cis-acting elements appear to contribute to transcription regulation. Based on these findings, the authors speculate that the amount of TFAM associated with nucleoid may differentially regulate transcription initiation and mtDNA replication [134].
The important role of TFAM in the coordination of mitochondrial transcription and replication has been confirmed using transgenic models. Loss of TFAM in mice results in mtDNA depletion leading to embryonic lethality [135]. Moderately elevated TFAM levels have resulted in elevated amounts of mtDNA transcripts, whereas its excessive overexpression has inhibited both mtDNA transcription and replication [136, 137]. It has recently been suggested that the nucleoid structure is a regulative factor for mitochondrial transcription: more dense mtDNA packaging mediated by excessive TFAM leads to lower accessibility of mtDNA to the transcription apparatus [138].
Transcription Termination
In addition to regulation of transcription initiation, mitochondrial transcription is also controlled at the termination step. Mitochondrial transcription initiated from the HSP2 generates a full-length RNA transcript, while transcription started from the HSP1 transcribes the 12S and 16S rRNA, tRNAVal, and tRNAPhe and terminates at the 3′ end of the tRNALeu(UUR) (Fig. 4.8) [139]. Specific binding of the mitochondrial transcription termination factor (mTERF1) to a 28-bp region at the 3′ end of the tRNALeu(UUR) promotes termination [140, 141].
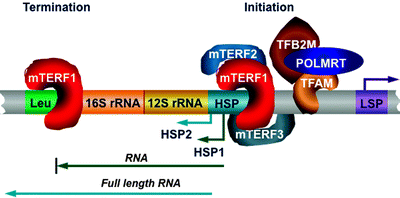
Fig. 4.8
Role of the mitochondrial transcription termination factor family members in mitochondrial DNA transcription. Mitochondrial transcription from the heavy-strand promoter (HSP) 2 generates a full-length RNA transcript, while transcription from the HSP1 transcribes the 12S and 16S rRNA, tRNAVal, and tRNAPhe and terminates at the 3′ end of the tRNALeu(UUR) (Leu). Binding of mitochondrial transcription termination factor (mTERF) 1 to the Leu is required for transcription termination. mTERF1 appears to bind simultaneously to both the termination region and initiation HSP1 site, resulting in the rDNA looping. Two other mTERF members may also interact with the initiation site: mTERF2 plays a role of transcription activator, while mTERF3 acts as a transcription repressor. Other abbreviations: LSP light-strand promoter; POLMRT mitochondrial RNA polymerase; TFAM transcription factor A of mitochondria; TFB2M transcription factor B2, mitochondrial
Two other members belonging to the mTERF family, mTERF2 and mTERF3, have been identified [142–144]. Highly homologous mTERF proteins contain conserved leucine zipper-like domains, well-known mediators of both protein–protein interactions, and DNA binding. The three-dimensional structure of mTERF3 has recently revealed a novel DNA-binding domain: multiple α-helices separated by loops, which can adopt a half-doughnut shape forming upon binding to DNA a half clamp [145].
Ability of the mTERF proteins to bind to DNA has been confirmed experimentally. mTERF1 has a high affinity to the tRNALeu(UUR) region and moderate affinity to the HSP1 and other regions in the D-loop [105]. mTERF2 and mTERF3 also display preferential binding to the HSP [146, 147]. Moreover, mTERF2 has been identified in isolated nucleoid preparations [148].
Importantly, mTERF1 appears to bind simultaneously to both the termination region and initiation HSP1 site, resulting in the rDNA looping [130]. Two other mTERF members may also interact with the initiation site (Fig. 4.8) [146]. However, it is currently unclear whether all three mTERF proteins assemble in a complex to bind to the HSP or mTERF1 forms a dimer with either mTERF2 or mTERF3 to promote mtDNA transcription. Depletion of mTERF2 has led to decrease in mitochondrial mRNA levels, while mTERF3 depletion has resulted in stimulation of mitochondrial transcription [146, 147]. Based on these findings, it has been suggested that mTERF2 plays the role of transcription activator, while mTERF3 acts as a transcription repressor. In addition, human mTERF1 contributes to mtDNA replication modulating replication pausing, highlighting further the coordination of mtDNA transcription and replication [105].
A termination site (H2 termination) of the long H2 transcript initiated from the HSP2 has been less characterized. A 22-bp A/T-rich region appears to represent the H2 termination region. ATAD3 and the leucine-rich pentatricopeptide-repeat containing protein (LRPPRC, also known as LPR130) can bind this region contributing to the H2 termination [149]. LRPPRC is essential for OXPHOS components encoded by mtDNA, and its deficiency results in reduced oxygen consumption associated with decrease in mRNA and tRNA levels [149–151]. Importantly, mutated LRPPRC has been found in Leigh syndrome French-Canadian patients displaying cytochrome oxidase deficiency [152].
Translation in Mitochondria
Mitochondrial mRNAs encoded by mtDNA are translated within the organelle on mitochondrial ribosomes [21]. Mitochondrial translation system evolved from the bacterial ancestor system: the catalytic characteristics of bacterial and mitochondrial ribosomes are similar, and bacterial and mitochondrial translation factors mediating initiation, elongation, and termination of protein synthesis are conserved [153, 154]. However, during almost two billion years of evolution, mitochondrial translation system has significantly changed compared to its bacterial counterpart, and some important differences have also evolved among different species [155–157].
Long polycistronic transcripts translated from mtDNA are processed to yield individual mitochondrial mRNAs, rRNA, and tRNAs. According to the tRNA punctuation model, specific folding of tRNAs generates substrates for mitochondrial RNases (e.g., RNase P), which promote excision of tRNAs to produce mRNAs and rRNAs [23]. Maturation of mRNAs includes the addition of a poly(A) tail to their 3′-termini, while tRNAs are maturated by the addition of CCA to their 3′-termini. These modifications are mediated by poly(A) polymerase (mtPAP) and tRNA nucleotidyltransferase, respectively [158, 159]. Moreover, mitochondrial mRNAs, unlike cytosolic mRNAs, do not have any modification of the 5′-termini. In addition, animal mitochondrial mRNAs lack significant 5′- and 3′-untranslated regions; thus, the start codon is typically located within three nucleotides of the 5′ end of the mRNA. The UAA stop codon of mitochondrial mRNA is enhanced by the posttranscriptional addition of a poly(A) tail.
Mitochondrial Ribosomes
All ribosomes are composed of two subunits: the small subunit promotes the decoding of mRNAs, whereas the large subunit contains the peptidyl transferase activity mediating peptide bond formation. The forming polypeptide is fed into the polypeptide tunnel, and the newly synthesized polypeptide leaves the ribosome from the polypeptide tunnel exit to be exposed to the hydrophilic milieu. The tunnel exit of ribosomes represents important regulatory site where various factors interact with the ribosome to govern the newly generated proteins [160]. However, in contrast to bacterial and yeast system, very little is currently known about human mitochondrial ribosomal tunnel exit.
The human mitochondrial ribosomes have a lower sedimentation coefficient, i.e., 55S, compared to bacterial 70S and cytosolic 80S counterparts [161]. However, they are larger than bacterial ribosomes based on their particle mass and physical dimensions [155, 162, 163]. The human mitochondrial ribosomes contain almost half the RNA found in bacterial ribosome, with a concomitant increase in the number of ribosomal proteins accounting for a significantly lower rRNA/protein ratio. The small 28S subunit of the human mitochondrial ribosome contains a 12S rRNA and over 30 proteins, whereas the large 39S subunit consists of a 16S rRNA and approximately 48 proteins [164]. These differences result in more porous and open structure with the absence of conventional E-site in mitochondrial ribosomes [161].
In contrast to the mitochondrial rRNA encoded by mtDNA, the mitochondrial ribosomal proteins (MRPs) are the products of nuclear genes [165, 166]. Gene map loci have been reported for the majority of human MRPs [167, 168]. The mapped genes are widely dispersed throughout the nuclear genome, suggesting that they were not transferred all together. Importantly, some of these genes overlap with chromosomal loci associated with various diseases, such as deafness, retinitis pigmentosa, and Usher syndrome [167].
Analysis of amino acid sequences of the majority of human MRPs revealed that most of them are highly homologous to the bacterial ribosomal proteins; although some homologues in small and large subunit are missing accounting for a net mass loss of over 200 kDa, this loss is highly compensated by the acquisition of more than 30 new proteins accounting for a net mass excess of about 870 kDa [161, 166]. Some of these new proteins appear to be multifunctional with extraribosomal functions. One of them, MRPS29 (known also as DAP3), is a GTP-binding protein located in the small subunit; together with MRPS30 (known also as PDCD9), it has previously been implicated in apoptosis [169–171]. Moreover, MRPS29 may mediate the 28S subunit binding to the MIM. Whether their roles for the mitochondrial ribosome are related to their functions in apoptosis remain to be determined. The precise locations of the majority of new MRPs are also unknown.
Translation Initiation and Elongation
In contrast to prokaryotic or eukaryotic cytosolic systems, the small 28S subunit is able to bind to mRNA tightly in the absence of initiating factors or tRNA. At least 400 nucleotides of the messenger template are minimally required for efficient binding to the ribosomal subunit. This may explain why the ATPase8 and ND4L transcripts, which are <300 nt, have to be part of a larger message [172].
Another important difference between mitochondrial and cytosolic translation is the former is initiated by a much reduced initiation complex, which appears to be composed by two initiation factors, mtIF2 and mtIF3 [173–175]. Mammalian mtIF2 belongs to a family of GTPases and exhibits strong structural and functional homology with E. coli IF-2. mtIF2 promotes fMET-tRNA binding to the 28S subunit in the presence of mRNA and GTP and appears functionally replaces both bacterial initiation factors, IF1 and IF2 [154]. Mitochondrial elongation factors, mtEF-Ts, mtEF-Tu, and mtEF-G1, sharing homology with the corresponding bacterial factors, mediate translation elongation in human mitochondrial ribosomes [176–179].
Similar to bacterial protein synthesis, protein synthesis mediated by mammalian mitochondrial ribosomes is sensitive to chloramphenicol but insensitive to cycloheximide. Chloramphenicol has been widely used to selectively inhibit mitochondrial protein synthesis without affecting the cytoplasmic protein synthesis machinery; conversely, cycloheximide is used to inhibit cytoplasmic protein synthesis with little effect on mitochondrial protein synthesis. The binding site for chloramphenicol can be modified by a mutation in the rRNA gene, resulting in chloramphenicol-resistant (CAPR) mutants [180].
Translation Termination
The standard stop codon UGA encodes Trp, whereas AGA and AGG serve as stop signals for mitochondrial transcripts MTCO1 and MTND6, the remaining 11 open reading frames terminate at UAA or UAG. Bacterial translation system utilizes two release factors (RFs) to terminate transcription: RF1 recognizes UAA/UAG, while RF2 recognizes UGA/UAA to mediate peptidyl-tRNA hydrolysis at corresponding stop codons [181, 182]. Two candidates for the role of human mtRF, related to bacterial RF1, have been identified. While biochemical analysis of mtRF1 has failed to reveal release activity with any codons, mtRF1a recognizes UAA/UAG, but is unable to recognize AGA/AGG [183, 184]. Given that AGA/AGG is directly preceded by a “U” nucleotide, it has recently been suggested that human mitochondria can utilize a −1 frameshift, which converts AGA/AGG in the standard UAA/UAG stop codons. Therefore, only single RF, such as mtRF1a, is needed for translation termination [185].
Finally, recycling of the human mitochondrial ribosome following translation termination is mediated by the mitochondrial ribosome recycling factor (mtRRF) acting in ensemble with mtIF3 and mtEF-G2 [186–189]. Interestingly, human mtRRF contains an N-terminal presequence for its targeting to mitochondria, which is not cleaved after its import to the organelle [186]. However, role of the mtRRF N-terminal region remains to be determined.
Mitochondrial DNA Repair
mtDNA Damage
mtDNA, similar to its nuclear counterpart, is a target for various damaging agents, including ionizing radiation, chemicals, environmental toxins, and pharmacological drugs. A negative charge on the inner surface of the MIM, generated by Δψm, results in tremendous accumulation and concentration (up to 1,000-fold) of lipophilic cations in mitochondria [190]. Many toxic chemicals represent positively charged lipophilic substances and therefore tend to accumulate in mitochondria affecting their function. Another factor contributing to mtDNA vulnerability is the lack of histones able to protect DNA. These factors explain significantly higher (tenfold or more) damaging of mtDNA by alkylating agents compared to that of nDNA [191].
mtDNA is particularly susceptible to oxidative damage caused by ROS generated during OXPHOS [192]. Importantly, in human cells, mtDNA damage is not only more extensive but also persists longer following OS than nDNA damage [193]. Various lesions, including base modifications, abasic sites, and single-strand breaks, are generated upon oxidative DNA damage [194]. Among them 8-oxoguanine (8-oxoG) is one of the most studied DNA lesions. 8-oxoG is a mutagenic lesion: its mispairing with adenine leads to a G–C to T–A transversion upon the subsequent round of replication. It has been demonstrated accumulation of 8-oxoG with age in both nDNA and mtDNA and to a greater extent in the latter [192, 195].
Mitochondrial DNA Repair Mechanisms
Despite the high vulnerability of mtDNA, it has been initially assumed that DNA repair is absent or very inefficient in mitochondria. Damaged mtDNA has been believed to be degraded, while undamaged copies have been used as templates for mtDNA synthesis. However, more recently, it has been shown the efficient repair of certain types of DNA lesions in mitochondria [196–199]. mtDNA repair mechanisms, including base excision repair (BER), mismatch repair, and recombinational repair, have been extensively investigated in Saccharomyces cerevisiae, and multiple yeast proteins implicated in these pathways have been identified [200–203]. Several mammalian mitochondrial proteins, which may contribute to various DNA repair mechanisms, have subsequently been identified; the growing protein list is summarized in Table 4.2 [198, 199, 204].
Table 4.2
Mammalian proteins potentially involved in mtDNA repair
Protein | Function |
|---|---|
APE1 (also known as APE, Apn1) | Apurinic/apyrimidinic endonuclease; BER |
OGG1 (MutM homologue) | 8-oxoguanine DNA glycosylase; BER |
UNG1 | Uracil DNA glycosylase; BER |
MtTGendo | DNA glycosylase; BER |
MYH (MutY homologue) | DNA glycosylase; BER |
Mlh1 (MutL homologue) | Molecular matchmaker; MMR |
Msh5 (MutS homologue) | Mismatch recognizing protein; MMR |
POLγ holoenzyme | DNA polymerase |
LIG3 | DNA ligase |
TFAM (also known as mtTFA) | mtDNA packaging |
mtSSB | ssDNA-binding protein |
EndoG | Endonuclease |
Rim1 (also known as SSB) | ssDNA-binding protein |
DUT1 | dUTPase |
MTH1 | Triphosphatase |
BER, the major mtDNA repair mechanism, targets oxidatively modified bases, including 8-oxoG and thymine glycol. A number of enzymatic activities promoting this repair pathway have been identified in human mitochondria (Table 4.2). It is initiated with recognition followed by removal of a damaged or inappropriate base by damage-specific DNA glycosylases, including uracil DNA glycosylase (UDG), 8-oxoG DNA glycosylase/AP lyase (OGG1, a homologue of E. coli MutM), thymine glycol glycosylase (NTH), and MYH glycosylase (homologue of E. coli MutY glycosylase). These enzymes cleave the N-glycosylic bond between the base and the sugar leaving an abasic site. This abasic site is recognized by apurinic/apyrimidinic nucleases, such as APE1 or APE2, which generate a 3′-OH and 5′-phosphate DNA break. POLγ extends the formed 3′-OH moiety, and DNA ligase, such as LIG3, joins DNA ends completing repair. In contrast to the short-patch and long-patch BER present in the nucleus, only the short-patch pathway, replacing a single damaged base, has been found in mitochondria [205]. Moreover, it has been demonstrated that mitochondrial proteins involved in BER are not freely soluble in the mitochondrial matrix, but are rather associated with the MIM [206].
In addition to BER, it has been reported that rat mitochondrial lysates can promote removal of GT and GG mismatches [207]. Moreover, proteomic analysis of mouse mitochondria has revealed the presence of Mlh1 and Msh5, mammalian homologues of E. coli mismatch repair proteins MutL and MutS, respectively [10]. These data suggest that mitochondria possess mismatch repair capacity; however, the role of this pathway in mtDNA maintenance remains to be determined.
Mitochondrial BER pathway can efficiently remove damaged bases; however, DNA double-strand breaks and cross-links can be repaired via homologous recombination and/or nonhomologous end joining mechanisms. Although emerging evidence suggests that human mitochondria possess homologous recombination activity, which has further been confirmed by proteomic identification of several proteins involved in this pathway, the extent and importance of mtDNA recombination repair remain a matter of open debate [199, 208–211]. Interestingly, in adult human heart, mtDNA is organized in complex catenated networks containing abundant recombination intermediates, Holliday junctions [82]. Similar mtDNA organization with multiple recombination junctions has been found in human and mouse brain but not in other tissues. Moreover, cardiac mtDNA recombination intermediates appear to correlate with mtDNA copy number [212]. Overexpression of TFAM and Twinkle in mice has led to elevated mtDNA copy number associated with a significant increase in recombination intermediates in the heart, brain, and skeletal muscles [82]. Based on these recent findings, it has been hypothesized that recombination mechanisms may play particularly an active role in mtDNA maintenance in the heart. In the myocardium, ROS originating from the highly active OXPHOS represent an abundant source of mtDNA damage [213]. Oxidatively modified nucleotides can be repaired via BER mechanisms; however, they can also lead to double-strand breaks, which require recombinational repair [214]. In addition to a protective role against oxidative mtDNA damage, ROS-induced recombination-dependent mtDNA replication associated with accelerated mitochondrial biogenesis may represent a myocardial response to increased energy demand.
Physiological Significance of mtDNA Repair
Modulation of mtDNA repair has been more readily addressed in yeast, whereas in humans, such studies are very limited. Targeting of overexpressed OGG1, a glycosylase that removes 8-oxoG, in HeLa mitochondria has significantly enhanced the resistance to OS [215–217]. However, Ogg1-deficient mice, although accumulate 10- to 20-fold higher 8-oxoG levels in mtDNA, display no mitochondrial dysfunction and develop and age normally [218–220]. Similarly, UDG knockout mice deficient in both mitochondrial and nuclear activities show no apparent mitochondrial defects under normal conditions, although they have increased postischemic brain injury [221]. Consistently, inhibition of S. cerevisiae UDG has not resulted in increased mutagenesis of mtDNA [222].
Growing evidence suggests a role for mtDNA repair in the pathogenesis of neurodegenerative diseases and carcinogenesis; however, the exact contribution of mtDNA repair pathways to these pathologies remains to be determined [204]. Lastly, age-dependent accumulation of oxidative damage in mtDNA in cardiomyocytes leading to various mutations and the role of mtDNA repair mechanisms in this process will be discussed in Chap. 13.
Mitochondrial Protein Biogenesis
Only 13 proteins are produced within the mitochondria, while the remaining ∼1,500 mitochondrial proteins are synthesized on cytosolic ribosomes and are imported into the organelle. Translocation of precursor proteins across the mitochondrial membranes relies on the presence of various mitochondrial targeting signals in their structure. Major targeting and sorting signals of mitochondrial precursor proteins are schematically shown in Fig. 4.9 [2, 223].
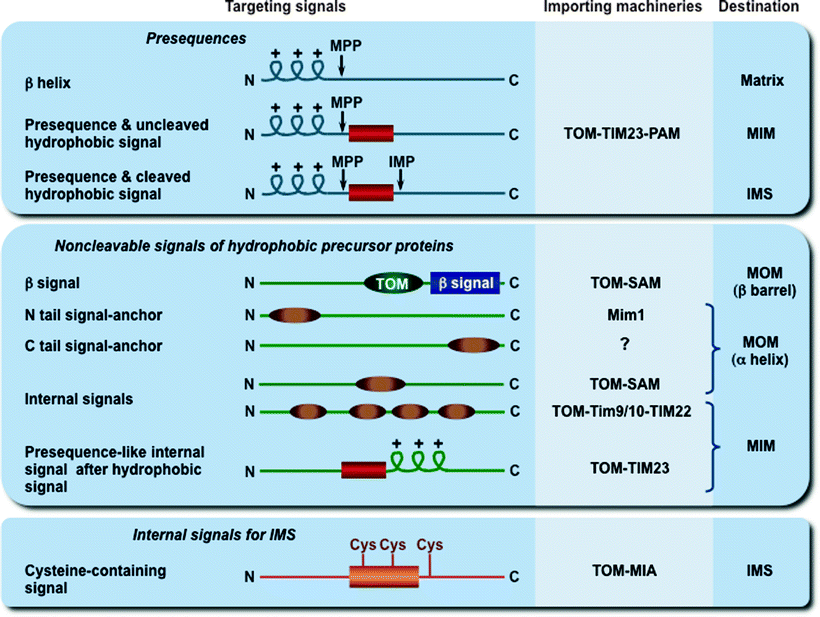
Fig. 4.9
Mitochondrial targeting and sorting signals. The three major types of cleavable and noncleavable signals, which target the precursor proteins to different importing and sorting machineries, are depicted. Presequences are typically removed after import by the mitochondrial processing peptidase (MPP), while hydrophobic sorting signals are removed by the inner membrane peptidase complex (IMP). The putative signal (TOM in dark green) for β-barrel preprotein targeting to the TOM complex is shown. IMS mitochondrial intermembrane space, MIA mitochondrial intermembrane space assembly, MIM mitochondrial inner membrane, Mim1 insertase of the mitochondrial outer membrane, MOM mitochondrial outer membrane, PAM presequence translocase-associated motor, SAM sorting and assembly machinery, TIM22 and TIM23 translocases of the MIM, Tim9/10 chaperone complex in the IMS, TOM translocase of the MOM
Structure and composition of major protein import machineries, translocase of the MOM (TOM) and two translocases of the MIM (TIM22 and TIM23), which promote mitochondrial protein import and sorting, have been discussed in Chap. 3. In this section, the mechanisms and dynamics of the main pathways implicated in the biogenesis of mitochondrial proteins will be overviewed.
Cleavable presequences represent the classical signals; they have a length of usually 15–50 amino acid residues, located at the N-terminus of precursor proteins, and form positively charged amphipathic α-helices. Most presequences are cleaved off by the mitochondrial processing peptidase (MPP) in the matrix, while some hydrophobic signals are removed by the MIM peptidase. All MOM proteins, most intermembrane space proteins, many multispanning MIM proteins, and a few matrix proteins contain noncleavable internal targeting signals, located at various positions within their molecules [2]. The exact targeting mechanisms of these internal signals are currently unknown.
It was initially thought that one common pathway, the presequence pathway, mediates import and assembly of all mitochondrial precursor proteins. However, it becomes clear that at least four major pathways are exploited for biogenesis of mitochondrial proteins, including the presequence pathway, the carrier protein pathway, the redox-regulated import pathway, and the β-barrel protein pathway [2, 223].
Presequence Pathway
The presequence pathway directs presequence-containing precursor proteins to the mitochondrial matrix and MIM. This pathway is mediated by the TOM and the TIM23–PAM (presequence translocase-associated motor) machineries in cooperation with the matrix motor and the ETC. Energy required for the process is provided by Δψm and ATP hydrolysis.
According to the binding chain model, the presequence of preprotein binds sequentially to subunits of the TOM complex: to Tom20–Tom22, then with assistance of Tom5 to the Tom40 channel, and finally to the intermembrane domain of Tom22 and Tom7 (Fig. 4.10) [224–228].
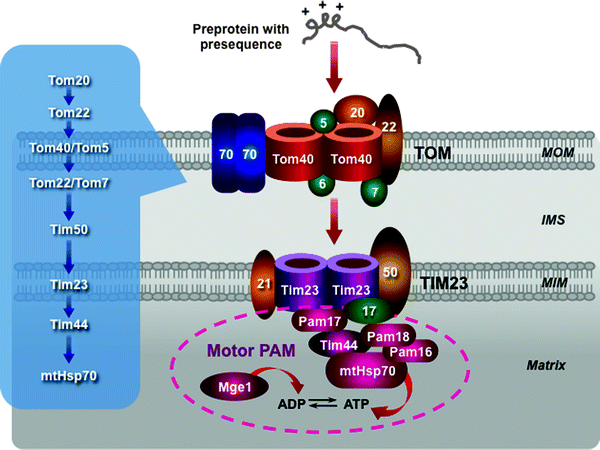
Fig. 4.10
Scheme of the presequence pathway. The translocase of the outer mitochondrial membrane (TOM) complex, the presequence translocase of the mitochondrial inner membrane (MIM) (TIM23 complex), and the presequence translocase-associated motor (PAM) are depicted. A sequential binding chain for presequence-containing precursor proteins is shown in the insert. See text for details. Other abbreviations: IMS intermembrane mitochondrial space, Mge1 mitochondrial GrpE-related protein 1, MOM mitochondrial outer membrane, mtHsp70 mitochondrial 70-kDa heat-shock protein
This chain of presequence binding events continues with the TIM23–PAM machinery. First, a preprotein translocated through the TOM complex is directly bound to Tim50, which mediates its binding to the intermembrane space domain of Tom22. Tim21 interacts with Tom22 to induce the preprotein release. Freed preprotein enters the channel-forming Tim23. Δψm activates Tim23 and generates an electrophoretic force facilitating translocation of the preprotein through the channel. Moreover, Tim21 interacts also with Complexes III and IV supporting the Δψm-driven entering of preproteins [229].
At this stage, the presequence pathway divides into two pathways, which are mediated by the lateral sorting or motor activity. The preproteins with hydrophobic sorting signals (the MIM proteins) are released within the MIM, while the majority of the preproteins are translocated into the mitochondrial matrix by the PAM machinery. The PAM subunits interact with the TIM23 complex: Pam17 binds to Tim23, while Pam18 binds to Tim17 [228, 230, 231]. The molecular chaperone heat shock protein 70 (mtHsp70) plays a key role in the ATP-consuming motor PAM. Tim44, in addition to preprotein binding, provides an ATP-sensitive site for mtHsp70 (Fig. 4.10) [232–236]. Tim44–mtHsp70 binds to and translocates preproteins into the matrix. This process requires ATP hydrolysis. Mge1, a nucleotide exchange factor, stimulates ATP cleavage cycle by releasing ADP from mtHsp70. J-domain-related chaperones Pam16 and Pam18 form a stable complex, which activates the ATPase activity of mtHsp70 [237, 238]. In contrast to Tim50 and Tim23, the PAM proteins do not require presequences for binding, recognizing the unfolded state of importing preproteins. Finally, the presequences are cleaved off by the mitochondrial processing peptidase (MPP) in the matrix to generate the mature matrix proteins. Several modifications of this principal dynamic model have recently been proposed [2].
Carrier Pathway
The carrier pathway promotes the import of the hydrophobic metabolite carrier proteins, to be integrated into the MIM. The precursor carrier proteins synthesized on cytosolic ribosomes are bound by molecular chaperones, belonging to the Hsp70 and Hsp90 families (Fig. 4.11). The TOM receptor subunit Tom70, containing binding sites for both the preproteins and the chaperones, binds the preprotein–chaperone complexes [239–241]. Due to the presence of several internal targeting signals, the carrier preproteins bind several Tom70 molecules. This prevents aggregation of the hydrophobic precursors and induces Tom70 oligomerization. The precursor is released in an ATP-dependent manner from the complex with the chaperones and is inserted into the TOM channel [239].
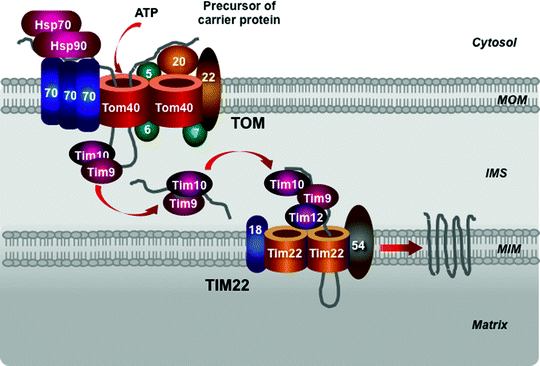
Fig. 4.11
Scheme of the carrier pathway. Cytosolic heat-shock protein (Hsp) 70 and Hsp90 chaperones, the translocase of the outer mitochondrial membrane (TOM complex), Tim9–Tim10 chaperone complex in the intermembrane mitochondrial space (IMS) and the translocase of the mitochondrial inner membrane (TIM22 complex) mediate import of the precursors of metabolite carriers of the mitochondrial inner membrane (MIM). See text for details. MOM mitochondrial outer membrane
An intermembrane space chaperone, the hexameric Tim9–Tim10 complex, binds the preprotein to direct it to the TIM22 complex [242–245]. In addition to the ability to bind the carrier preproteins, the Tim9–Tim10 complex can also bind other MIM noncleavable preproteins and the MOM β-barrel proteins. A homologous hexameric chaperone complex, formed by Tim8–Tim13, can also transfer the hydrophobic preproteins within the intermembrane space [246, 247]. The Tim9–Tim10 complex appears to play the main role in this process and is essential for cell viability, while the Tim8–Tim13 is dispensable for yeast growth. Interestingly, a missense mutation in the TIM8 gene has resulted in Mohr–Tranebjaerg syndrome, also known as deafness–dystonia syndrome [248].
Redox-Regulated Import Pathway
This pathway is mediated by the mitochondrial intermembrane space assembly (MIA) machinery to import small proteins containing cysteine motifs and forming intramolecular disulfide bonds. Interestingly, both subunits of the Tim9–Tim10 complex also contain two disulfide bonds, which are essential for the complex integrity [245]. Oxidoreductase Mia40 represents a key subunit of the MIA apparatus [249–251]. The precursors cross the MOM in the reduced state without disulfide bonds, which are stabilized in the cytosol by Zn2+, most likely through the TOM channel (Fig. 4.12) [243, 252–254]. In the intermembrane space, Mia40 binds to the redox-active cysteine–proline–cysteine motifs of the preprotein and forms transient mixed disulfide bonds with the preprotein [255–258]. The sulfhydryl oxidase Env1 cooperates with Mia40 in the oxidation of the preproteins. Env1 promotes the reoxidation of Mia40 cysteines transferring electrons to cytochrome c or directly to O2 [259–261]. A third known player of the MIA system, the cysteine-rich Hot13 binds Zn2+, keeps Mia40 in a Zn2+-free form, facilitating thereby its oxidation by Env1 [262, 263]. Interestingly, the Mia40 and Env1 precursors are also imported through this pathway [264].
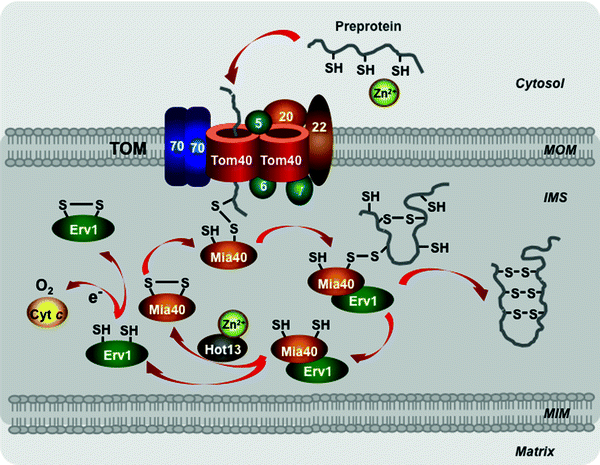
Fig. 4.12
Scheme of the redox-regulated pathway. Major players involved in import of the precursor proteins with cysteine motifs to the mitochondrial intermembrane space are shown. See text for details. Abbreviations: Cyt c cytochrome c, Erv1 mitochondrial oxidase “essential for respiration and viability” 1, Hot13 protein “helper of Tim” 13, IMS intermembrane mitochondrial space, Mia40 mitochondrial oxidoreductase “mitochondrial IMS assembly” 40, MIM mitochondrial inner membrane, MOM mitochondrial outer membrane, TOM translocase of the outer mitochondrial membrane
Biogenesis of the MOM Proteins
The MOM proteins belong to two main types of membrane proteins: α-helical proteins are inserted into the membrane with hydrophobic α-helical regions, whereas β-barrel proteins anchored to the membrane with multiple β strands. Signal-anchored proteins Tom20 and Tom70, many proteins implicated in mitochondrial dynamics, and cell death are α-helical proteins, whereas the central, channel-forming component of the TOM complex (Tom40) and the abundant MOM proteins (VDAC and Sam50) are well-known examples of mitochondrial β-barrel proteins. Biogenesis of the mitochondrial β-barrel proteins is currently better characterized.
The mitochondrial β-barrel preproteins are first translocated via the TOM complex into the mitochondrial intermembrane space (Fig. 4.13). Then the Tim9–Tim10 and Tim8–Tim13 complexes transfer the precursors to the sorting and assembly machinery (SAM) embedded in the MOM [265–268]. Two SAM subunits, the channel-forming Sam50 and the receptor Sam35, cooperate in the insertion of the β-barrel preproteins into the MOM [265, 269–272]. The third SAM component, Sam37, appears to mediate the release of β-barrel preproteins from the SAM machinery through yet undetermined mechanism [273].
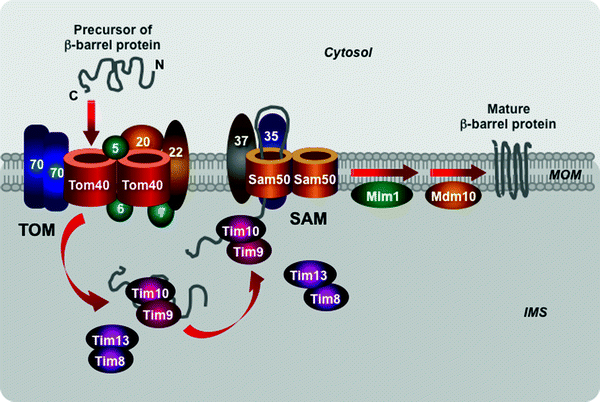
Fig. 4.13
Scheme of import of β-barrel proteins into the mitochondrial outer membrane. The translocase of the outer mitochondrial membrane (TOM) complex mediates translocation of β-barrel preproteins into the intermembrane mitochondrial space (IMS). The IMS translocase of the inner mitochondrial membrane Tim9–Tim10 and Tim8–Tim13 complexes transfers the preproteins to the sorting and assembly machinery (SAM). The SAM subunits Sam50 and Sam35 mediate insertion of the precursors into the mitochondrial outer membrane (MOM). See text for details. Other abbreviations: Mdm10 protein “mitochondrial distribution and morphology” 10, Mim1 mitochondrial import protein 1
Two additional MOM proteins, Mdm10 and Mim1, interact with the SAM complex, contributing to sorting and assembly of mitochondrial β-barrel proteins (Fig. 4.13). Mdm10, which is also a component of the MDM/mitochore complex, contributes to the assembly of the channel-forming Tom40 with the receptor Tom22 subunit and small Tom proteins, Tom5, Tom6, and Tom7, into mature TOM complex [274, 275]. The integral MOM protein Mim1 forms homooligomers that mediate the integration and assembly of signal-anchored components of the TOM complex, Tom70 and Tom20 [276–278]. Mim1 appears to be able to modulate the SAM complex function contributing to biogenesis of β-barrel as well as some α-helical proteins.
Various pathways appear to be involved in the targeting of α-helical proteins to the MOM; however, the major players and the exact molecular mechanisms of their biogenesis are the subject of ongoing research.
Mitochondrial Lipid Biogenesis
Biogenesis of the mitochondrial membranes relies on a highly coordinated synthesis, import, and assembly of proteins and phospholipids. However, in contrast to biogenesis of mitochondrial proteins, the mechanisms of mitochondrial phospholipid biosynthesis and trafficking are less characterized. CL structure and its essential role as a critical integrator of numerous mitochondrial processes ranging from the structural organization of mitochondrial membrane and ATP synthesis to metabolite and protein transport and apoptosis have been discussed in Chap. 3. This section will focus mainly on CL biosynthesis and recently discovered regulators of mitochondrial phospholipid biogenesis.
Mitochondrial Phospholipids
All main types of membrane phospholipids characteristic to all cellular membranes, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidic acid (PA), are present in the mitochondrial membranes. However, mitochondrial membranes also contain a unique phospholipid, cardiolipin (CL), located predominantly in the MIM. The most abundant PC and PE account for ∼40% and ∼30%, respectively, CL and PI account for 10–15%, while PA and PS comprise ∼5% of the total mitochondrial phospholipids [279–281]. Phosphatidylglycerol (PG), PG phosphate (PGP), and cytidine diphosphate-diacylglycerol represent intermediates of phospholipid biosynthesis. In contrast to other cellular membranes, the mitochondrial membranes contain only trace amounts of sphingolipids and sterols [282].
Phospholipids, depending on their shapes, can be divided into two groups: bilayer- and non-bilayer-forming phospholipids [283]. Bilayer-forming phospholipids, such as PC, PI, and PS, have cylindrical shapes with almost equivalent diameters of hydrophilic head groups and the hydrophobic FA portions, favoring bilayer formation (Fig. 4.14). CL, PE, and PA are non-bilayer-forming phospholipids, which are characterized by more conical shapes with smaller head group diameters. Within bilayer membranes, non-bilayer phospholipids induce membrane curvature or form a specific microdomain with the exposed hydrophobic domains (Fig. 4.14). This feature of non-bilayer phospholipids can explain the important role of CL in various mitochondrial processes [283].
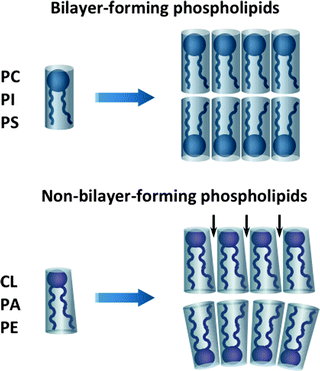
Fig. 4.14
Bilayer- and non-bilayer-forming phospholipids of mitochondrial membranes. Bilayer-forming phospholipids, phosphatidylcholine (PC), phosphatidylinositol (PI), and phosphatidylserine (PS), have cylindrical shapes, favoring bilayer formation. Non-bilayer-forming phospholipids, cardiolipin (CL), phosphatidylethanolamine (PE), and phosphatidic acid (PA), are characterized by more conical shapes with smaller head group diameters. Within bilayer membranes, non-bilayer phospholipids induce membrane curvature or form a specific microdomain with the exposed hydrophobic domains (marked with arrows)
Membrane curvature at cristae tips and cristae junctions may recruit the non-bilayer-forming phospholipids CL and PE. Mitochondrial membrane scaffolding proteins may contribute to assembly of the CL- and PE-containing microdomains. The evolutionary conserved proteins, prohibitins, assemble into ring complexes in the MIM and may form a scaffold for the CL- and PE-enriched domains within the MIM [284–286].
Mitochondrial Phospholipid Biosynthesis
CL, PE, PG, and PA are synthesized in mitochondria, while PC, PI, and PS are synthesized predominantly in the endoplasmic reticulum (ER) and are subsequently imported in the organelle. The molecular mechanisms implicated in the biosynthesis of mitochondrial phospholipids are well characterized in S. cerevisiae. Major enzymes involved in this process are evolutionary conserved in mammalian cells.
The first step in the biosynthesis of all phospholipids is initiated by glycerol-3-phosphate (G3P) acyltransferases (GPATs) that catalyze the acylation of the sn-1 position of G3P to produce lyso-PA (Fig. 4.15). In mammals, GPATs are found in various organelles, including mitochondria [287]. Multiple lyso-PA acyltransferases promote conversion of lyso-PA into PA, a key intermediate of two distinct branches of phospholipid biosynthesis. One pathway generates cytidine diphosphate-diacylglycerol (CDP-DAG) catalyzed by CDP-DAG synthase (Cds1) to produce eventually PG, CL, PI, PS, and PE [288]. In another pathway, the phosphatase Php1 converts PA to DAG, which in turn is utilized in the so-called Kennedy pathway to generate PE and PC [289, 290].
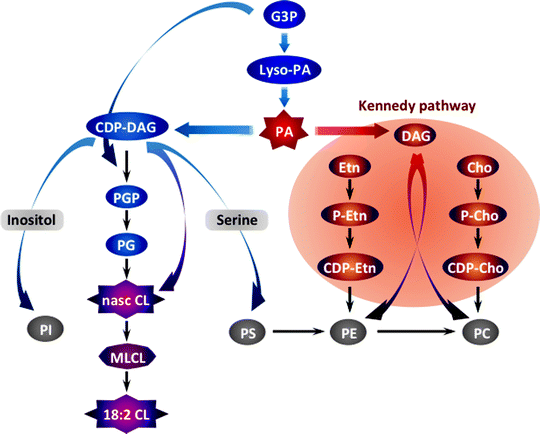
Fig. 4.15
Schematic summary of mitochondrial phospholipid biosynthesis. The biosynthesis of all phospholipids is initiated by the glycerol-3-phosphate (G3P) acyltransferase-mediated acylation of G3P to produce lysophosphatidic acid (lyso-PA). Multiple lyso-PA acyltransferases promote conversion of lyso-PA into phosphatidic acid (PA), a key intermediate of two distinct branches of phospholipid biosynthesis. One pathway generates cytidine diphosphate (CDP)-diacylglycerol (DAG) catalyzed by CDP-DAG synthase to produce eventually phosphatidylglycerol (PG), cardiolipin (CL), phosphatidylinositol (PI), phosphatidylserine (PS), and phosphatidylethanolamine (PE). In another pathway, the phosphatase Php1 converts PA to DAG, which in turn is utilized in the so-called Kennedy pathway to generate PE and phosphatidylcholine (PC). Other abbreviations: Cho choline, Etn ethanolamine, MLCL monolysocardiolipin, P-Cho phosphorylcholine, P-Etn phosphorylethanolamine, PGP phosphatidylglycerophosphate
In mammals, PS formed in the ER is imported into mitochondria, where it is converted into PE, which in turn is exported to other organelles [291]. This route represents the main biosynthetic pathway accounting for the majority of cell membrane glycerophospholipids [282].
CL precursor, PA, is formed in the MOM and then migrates to the MIM, where in four enzyme-catalyzed steps, the nascent tetra-acyl CL, not enriched for linoleate, is synthesized [292, 293]. This newly synthesized CL is remodeled to the linoleate-enriched cardiac 18:2 CL by not completely understood mechanism. Two mitochondrial enzymes, monolysocardiolipin acyltransferase and tafazzin, and one ER enzyme, acyl-Coenzyme A, have been proposed to mediate CL remodeling [292, 294–296].
Although the main precursors of CL biosynthesis, PA and CDP-DAG, and enzymes implicated in the process are localized in the mitochondrial membranes, mitochondria may also use extrinsic sources of precursors for its formation. It is unclear how newly synthesized CL is redistributed within the mitochondrial membranes.
In the human heart, up to 80% of acyl side chains in CL are symmetrical linoleic acid chains (see Chap. 3) [297]. Such unique molecular structure contributes to interactions between adjacent CL molecules essential to the formation of CL-containing microdomains in the MIM, which in turn facilitates proton transfer during ATP synthesis by the OXPHOS complexes [298, 299].
The physiological importance of CL biosynthesis and remodeling has been highlighted by the discovery of mutations in the TAZ1 gene encoding the defective CL-remodeling enzyme, tafazzin, which causes the Barth syndrome [296, 300, 301]. This X-linked recessive disease is characterized by cardiomyopathy, neutropenia, and increased blood levels of 3-methylglutaconic acid [302, 303]. Consistently, alterations in CL biosynthesis have led to impaired mitochondrial function associated with acute myocardial I/R, heart failure, aging heart, and diabetes in experimental and clinical settings [301, 304, 305].
Mitochondrial Phospholipid Traffic
Phospholipid trafficking from the ER to mitochondria as well as within the organelle between the MOM and MIM appears to be mediated by non-vesicular pathways through close membrane contacts [306]. Specific fractions of the ER closely associated with mitochondria and referred to as mitochondria-associated membranes (MAM) are implicated in this process [307–311]. Protease sensitivity of phospholipid transport from MAMs to mitochondria suggests the involvement of membrane proteins in this process [312, 313]. Although multiple mammalian proteins have been suggested to play a role in MAM-mitochondria tethering, direct evidence for their involvement in phospholipid transport has not yet been demonstrated [314, 315].
Similarly, mechanisms underlying trafficking of newly imported or synthesized phospholipids within mitochondria are largely unknown. Mitochondrial contact sites may be involved, similar to protein import, in phospholipid trafficking between the MOM and MIM [309, 316]. Moreover, it has been demonstrated that mitochondrial creatine kinase and nucleotide diphosphate kinase can activate CL translocation between liposomes; however, the physiological significance of this finding is unclear [317].
PGC-1α: A Central Regulator of Mitochondrial Biogenesis
A variety of mitochondrial proteins is encoded by both nuclear and mitochondrial genomes; therefore, finely tuned coordination between expression of a subset of nuclear and mitochondrial genes is of a great importance. Nuclear–mitochondrial communications are orchestrated by an elaborate interplay between a plethora of transcription factors and transcription coregulators [11–14, 19]. The transcription factors target specific genes mediating main changes in their expression. Action of these factors is modulated by transcription coregulators, which sense metabolic changes and translate them into the fine-tuning of gene expression [11, 12, 318, 319].
The central role of nuclear-encoded transcription factors TFAM, TFB1M, and TFB2M in regulation replication, transcription, and maintenance of mtDNA has already been discussed in this chapter. Other transcription factors implicated in the control of mitochondrial biogenesis include the nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2), estrogen-related receptor α (ERRα), peroxisome-proliferator-activated receptors (PPARs), cAMP response element-binding protein (CREB), initiator element-binding factor (YY1), Sp1, and many others [19]. The peroxisome-proliferator-activated receptor γ coactivator-1 (PGC-1) family of transcription coactivators orchestrates this highly complex regulatory circuitry playing a role of a nodal regulator of mitochondrial biogenesis and respiratory function (Fig. 4.16) [13, 320, 321].
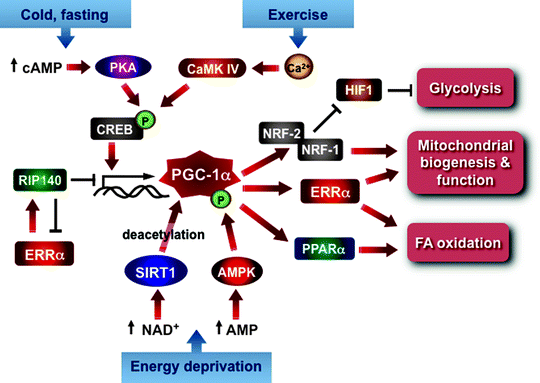
Fig. 4.16
PGC-1α as a central regulator of mitochondrial biogenesis and function. The scheme summarizes major positive and negative signals converging at PGC-1α, which orchestrates the highly complex transcriptional circuit involved in the regulation of mitochondrial biogenesis and function. See text for details. Abbreviations: AMPK AMP-activated protein kinase, CaMK IV calcium/calmodulin-dependent protein kinase 4, cAMP cyclic AMP, CREB cyclic AMP response element-binding protein, ERRα estrogen-related receptor alpha, HIF1 hypoxia-inducible factor 1, NRF nuclear respiratory factor, PKA cAMP-dependent protein kinase, PPARα peroxisome-proliferator-activated receptor alpha, RIP140 receptor-interacting protein 140, SIRT1 deacetylase 1
PGC-1 Family
The PGC-1 family is a small family of structurally homologous transcriptional coactivators, which is comprised of three members PGC-1α, PGC-1β, and PGC-1α-related coactivator (PRC) (Fig. 4.17). PGC-1α, the founder member of the family, was initially identified due to its interaction with PPARγ and its induction during adaptive thermogenesis in brown fat [322]. PGC-1α contains the N-terminal transcriptional activation domain, adjacent nuclear hormone receptor coactivator signature motifs LXXLL, central proline-rich region, and the C-terminal arginine/serine rich (R/S) and RNA recognition motifs (Fig. 4.17).

Fig. 4.17
Scheme of the molecular structure of the PPARγ coactivator 1 (PGC-1) family transcriptional coactivators. The N-terminal transcriptional activation domain (AD), adjacent nuclear hormone receptor coactivator signature motifs LXXLL, central proline-rich region (PRR), host cell factor binding motif (HCF), and the C-terminal arginine/serine rich (R/S) and RNA recognition motifs (RRM) are schematically shown. PRC PGC1-related coactivator
PGC-1α does not have histone-modifying activity; however, it binds multiple coactivator complexes with chromatin-remodeling capabilities, such as CBP/p300, GCN5, SRC-1, and SRC-3. Moreover, PGC-1α interacts with increasing number of transcription factors and nuclear hormone receptors, which control mitochondrial ETC, such as NRF1 and NRF2α, ERRα, MEF2C, and YY1, or FA oxidation, such as PPARα and ERRα (Fig. 4.16). NRF1 and NRF2α (also known as GABPα) act downstream from ERRα regulating the OXPHOS gene expression [18, 323]. ERRα in cooperation with ERRγ mediates the expression of genes implicated in adaptive thermogenesis as well as multiple metabolic genes essential for myocardial function [18, 324]. During adipogenesis, ERRα can also activate receptor-interacting protein 140 (RIP140) expression to suppress PGC-1α-dependent mitochondrial function and biogenesis [325, 326]. In addition, the nuclear receptor corepressor RIP140 interacts with PGC-1α and mediates negative control of the ERRα- and NRF1-dependent expression of the CIDEA gene, implicated not only in metabolism but also in cell death response [327]. Therefore, PGC-1α integrates both positive and negative transcriptional regulatory signals affecting mitochondrial biogenesis and function (Fig. 4.16).
A closely related PGC-1α homologue, PGC-1β (also known as PERC), displays a high degree of sequence similarities along its entire length, but it lacks R/S domain (Fig. 4.17) [328, 329]. Both PGC-1α and PGC-1β are highly expressed in tissues with high oxidative capacity and elevated mitochondria content, such as heart, skeletal muscle, brown adipose, and kidney; however, unlike PGC-1α, PGC-1β is not induced in brown fat upon cold exposure [328, 329]. Similar to PGC-1α, PGC-1β interacts with NRF-1 and activates expression of nuclear genes involved in regulation of mitochondrial biogenesis [330, 331].
The third PGC-1 family member, PRC, contains all characteristic motifs suggesting the functional similarities with other family members [332]. Accordingly, PRC interacts with transcription factors NRF1, ERRα, and CREB involved in control of mitochondrial biogenesis [332–334]. It trans-activates various nuclear genes essential for mitochondrial function, such as cytochrome c, TFAM, TFB1M and TFB2M, and 5-amino-levulinate synthase [332, 335].
Regulation of PGC-1α
Among PGC-1 family members, regulation of PGC-1α has been the most extensively studied. Both PGC-1α expression and its activity are finely tuned in response to a variety of external stimuli. The molecular mechanisms, which control PGC-1α expression, vary also in different tissues.
Transcriptional Regulation
Transcription of the PGC-1α gene is induced in response to conditions that demand elevated mitochondrial ATP production or thermogenesis, such as exercise, cold exposure, and fasting. Multiple pathways are implicated in exercise-induced PGC-α expression in skeletal muscle. In response to exercise, Ca2+ signaling activates Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) and calcineurin A (CnA). CaMKIV induces CREB binding to the CRE site in the PGC-1α promoter, whereas CnA activates the myocyte enhancer factor 2C and 2D (MEF2C and MEF2D, respectively) that bind a MEF2 response element in the PGC-1α promoter promoting PGC-1α transcription (Fig. 4.18) [336–340].
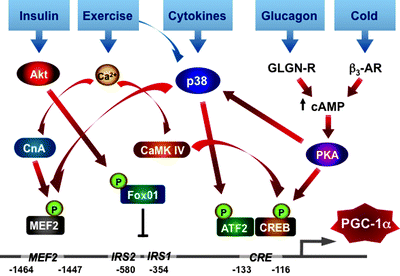
Fig. 4.18
Schematic summary of transcriptional regulation of PPARγ coactivator 1α (PGC-1α). Diverse physiological signals induce various signaling pathways, which phosphorylate and modulate activities of several transcription factors, such as cAMP response element-binding protein (CREB), activating transcription factor 2 (ATF2), forkhead box class-O (FoxO1), and myocyte enchancer factor 2 (MEF2). These factors interact with multiple cis-regulatory elements within the PGC-1α promoter and regulate PGC-1α expression. β 3 -AR β3-adrenergic receptor, CaMKIV calmodulin-dependent protein kinase IV, CnA calcineurin A, CRE cAMP response element, GLCN-R glucagon receptor, IRS insulin response element, p38 p38 mitogen-activated protein kinase, PKA protein kinase A. See text for details
Activation of the mitogen-activated protein kinase p38 MAPK represents another regulatory mechanism induced in muscle after exercise. Exercise-activated p38 MAPK induces PGC-1α transcription through the engagement of activating transcription factor 2 (ATF2) and MEF2 (Fig. 4.18) [341, 342]. This pathway can also be activated by cytokines.
Cold exposure induces the β3-adrenergic receptor (β3-AR) in brown fat activating cAMP signaling and resulting subsequently in upregulation of PGC-1α expression (Fig. 4.18) [343, 344]. This mechanism involves activation of PKA, which phosphorylates CREB, leading to its binding to PGC-1α promoter. In addition, β3-AR–PKA pathway can also induce p38-ATF2-mediated activation of PGC-1α transcription [345].
In response to fasting, pancreatic hormone glucagon induces cAMP–PKA–CREB-mediated activation of PGC-1α expression in liver (Fig. 4.18) [337]. Upregulation of PGC-1α transcription contributes to activation of multiple genes encoding gluconeogenic enzymes. Similar to skeletal muscle and brown fat, the fasting–glucagon–PKA pathway in liver can engage p38-ATF2 activation of PGC-1α transcription [346].
Insulin signaling pathway has also been implicated in regulation of PGC-1α expression in muscle (Fig. 4.18). Importantly, insulin appears to be able to mediate both activation and inhibition of PGC-1α transcription; however, the precise mechanisms of this pathway are poorly understood [347, 348]. Insulin can activate Akt to phosphorylate forkhead box class-O factor (FoxO1), leading to its dissociation from PGC-1α promoter and abrogating PGC-1α transcription. Unfortunately, control of PGC-1β transcription currently is much less characterized. However, it has been reported that interferon γ and interleukin-4 induce PGC-1β expression through STAT1 and STAT6, playing a role in host defense [349, 350]. Moreover, in activated macrophages, PGC-1β transcription, similar to PGC-1α, can be activated by cytokines [350].
Posttranscriptional Regulation
PGC-1α is also extensively regulated at the posttranscriptional level by a variety of modifications, including phosphorylation, acetylation, ubiquitination, methylation, and O-linked N-acetylglucosaminylation (O-GlcNAcylation). Numerous sites in PGC-1α are phosphorylated by AMPK, Akt, p38 MAPK, and glycogen synthase kinase 3β (GSK3β) affecting its activity and/or stability [321, 351]. In addition to activation of PGC-1α expression, AMPK stimulates its cotranscriptional activity by phosphorylating threonine-177 and serine-538 [352]. Similarly, the insulin-activated Akt not only downregulates PGC-1α transcription but also inhibits its activity by phosphorylating serine-570 [348, 353]. Moreover, the feeding–insulin–Akt signaling stabilizes Cdc2-like kinase 2, Clk2, which in turn phosphorylates SR region in PGC-1α to inhibit its activity and to downregulate thereby gluconeogenesis [354].
In response to cytokines in muscle, p38 MAPK phosphorylates threonine-262 and threonine-298 and serine-265, leading to PGC-1α stabilization and activation [355–357]. In addition, it also attenuates the inhibitory binding of the corepressor, p160 myb-binding protein to PGC-1α increasing its coactivator potential [358]. GSK3β also mediates the inhibitory phosphorylation of PGC-1α at threonine-295 in response to OS [359]. In this case, phosphorylated PGC-1α is targeted to proteosomal destruction in the nucleus; however, the functional significance of this event remains to be determined.
PGC-1α is acetylated at multiple sites along the whole molecule leading to inhibition of its cotranscriptional activity. Two enzymes, which act as sensors of the cellular energy status, are responsible for PGC-1α acetylation: the histone acetyltransferase GCN5 and the deacetylase silence information regulator 1 (SIRT1) [360].
Although it has been reported that PGC-1α can interact with various acetyltransferases, such as GCN5, p300, SRC-1, and SRC-3, only the former has been demonstrated to acetylate and inhibit PGC-1α cotranscriptional activity in vivo [336, 361–363]. It is important to note that SRC-3 can facilitate GCN5 ability to acetylate and inhibit PGC-1α. Under energy abundant conditions, the expression of GCN5 and SRC-3 is upregulated resulting in PGC-1α acetylation and inhibition of its activity, whereas in situations when cellular energy stores are reduced, GCN5 and SRC-3 expression is downregulated leading to stimulation of PGC-1α activity [363]. Although it is less known about posttranslational regulation of PGC-1β activity, it has recently been demonstrated that its cotranscriptional activity is also inhibited by GCN5-promoted acetylation [362].
The NAD+/NADH ratio regulates deacetylase activity of SIRT1 underlying its role in sensing the cellular energy status. Under energy-demanding conditions, such as exercise, fasting, or OS, increased NAD+/NADH ratio stimulates SIRT1 activity, which in turn deacetylates and activates PGC-1α to boost mitochondrial ATP production [364–366]. Consistent with this role, SIRT1 knockdown leads to downregulation of PGC-1α-activated gluconeogenesis, while SIRT1 overexpression results in activation of gluconeogenesis in mouse liver [360, 367].
AMPK not only phosphorylates and activates PGC-1α directly, but it also increases NAD+ levels stimulating SIRT1 activity and thereby activating PGC-1α indirectly [365]. Interestingly, AMPK-mediated phosphorylation of PGC-1α is essential for its deacetylation by SIRT1 interlinking these posttranslational modifications.
Ubiquitin-mediated proteolysis represents an additional regulatory mechanism of cellular PGC-1α levels. It has recently been demonstrated that Skp1/cullin/F-box-cell division control 4 (SCFCdc4) functions as PGC-1α-specific E3 ubiquitin ligase [368]. Given the stabilizing role of p38 MAPK-mediated phosphorylation of PGC-1α surprisingly, PGC-1α phosphorylation by both GSK3β and p38 MAPK appears to facilitate SCFCdc4-mediated targeting of PGC-1α for proteosomal destruction.
Finally, PGC-1α is activated by the protein arginine methyltransferase 1 (PRMT1)-mediated methylation at arginine-665, arginine-667, and arginine-669 [369]. In addition, PGC-1α serves also as a substrate for O-GlcNAc transferase that attaches O-GlcNAc group to serine-333 [370]. The functional significance of these modifications remains to be determined.
In Vivo Functions of PGC-1 Family Members
Despite the functional similarities between PGC-1 coactivators, PGC-1α deficiency downregulates the thermogenic gene expression, but does not inhibit brown fat differentiation, while PGC-1β silencing abolishes increase in mitochondria number during brown fat differentiation [371]. Moreover, overexpression of the two factors in skeletal muscle suggests that they appear to mediate distinct differentiation programs of muscle fiber types [372, 373]. In contrast to PGC-1α and PGC-1β, expression of PRC in various tissues does not correlate with mitochondrial abundance [332]. Furthermore, unlike PGC-1α expression, expression of PRC is only slightly induced in brown fat upon cold exposure. Importantly, PRC mRNA and protein levels are significantly induced in proliferating cells or upon serum stimulation of quiescent cells suggesting a role in the integration of mitochondrial component expression with the cell proliferation [332, 333].
Transgenic mouse models confirm the central role of PGC-1α and PGC-1β in orchestrating the expression of multiple genes essential for mitochondrial biogenesis. Surprisingly, PGC-1α- and PGC-1β-deficient mice are viable and have normal mitochondrial morphology and function suggesting that they are not required for normal development and mitochondrial biogenesis or maintenance [374–378]. Importantly, however, both PGC-1α and PGC-1β knockouts display some alterations of mitochondrial function under stress conditions, such as deficiencies in adaptive thermoregulation, modest downregulation in respiratory gene expression, and reduced myocardial mitochondria number and ATP production in response to physiologic stress [374–379]. Interestingly, increased expression of PGC-1α found in brown fat of PGC-1β-deficient mice is not able to compensate for the loss of PGC-1β confirming that two PGC-1 family members have nonoverlapping functions.
Recently generated PGC-1α/PGC-1β double knockout mice, although viable at birth, have died of HF within 24 h with no survivals beyond 14 days. Importantly, multiple mitochondrial abnormalities have been detected in the failing hearts of the double knockouts, including significant decrease in mitochondrial size and number, reduction in cristae, and the appearance of internal vacuoles [380]. Expression of several genes encoding proteins implicated in FAO and OXPHOS has also been markedly downregulated in these double knockouts. This finding highlights a complementary role of these coactivators in postnatal heart development. It is important to note that PRC might compensate the absence of PGC-1α and PGC-1β during embryogenesis of double knockout mice [380].
PRC silencing using short hairpin RNAs (shRNAs) or siRNAs has resulted in significant downregulation of components of complexes I, II, and IV, leading to reduced mitochondrial ATP production [381, 382]. In addition to the OXPHOS components, gene array analysis has also demonstrated downregulation of the genes encoding subunits of mitochondrial protein import machinery [381]. Thus, silencing of PRC leads to multiple mitochondrial defects resembling those observed in mouse knockouts of genes encoding essential mitochondrial proteins.
PGC-1-Mediated Regulatory Circuitry in the Heart
It is well known that after birth the myocardium switches from glucose and lactate oxidation to mitochondrial FA oxidation. PGC-1α and PGC-1β are needed and sufficient for this dramatic shift in cardiac fuel utilization. During the early postnatal period, PGC-1α and PGC-1β expression is significantly upregulated, accompanying by a burst in cardiac mitochondrial biogenesis [16, 380]. Accordingly, PGC-1α overexpression in cardiomyocytes induces mitochondrial biogenesis.
Both PGC-1α and PGC-1β play also essential role in the adult heart. Loss of PGC-1α or PGC-1β leads to a modest downregulation in FAO and reduced cardiac reserve with dobutamine treatment [376, 378–380, 383]. Chronic PGC-1α overexpression in the myocardium results in a severe but reversible cardiomyopathy accompanied by significant mitochondrial defects [384].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


