(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Both clinical studies and animal models have shown that mitochondria are at the center of the pathophysiology and pathogenesis of the failing heart since mitochondrial-based oxidative stress together with cell death and cardiac bioenergetic dysfunction is profoundly implicated in the progression of heart failure (HF). The evidence available on the role that mitochondrial defects play in the development of HF and potential new approaches to therapeutic targeting of mitochondrial bioenergetic, biogenetic, and signaling abnormalities will be discussed in this chapter.
Introduction
Heart failure (HF) is a pathophysiologic state where the heart is unable to meet the metabolic requirements of the body. A chronic disorder, HF is the principal cause of hospitalization in patients over 65 years of age. It has a progressive clinical course resulting in high morbidity and mortality and economically poses a tremendous burden for the health care delivery system.
Taking advantage of the remarkable fusion recently achieved by genetics and biochemistry in molecular biology, mitochondrial research is accelerating its application to cardiovascular pathologies. The essential role that mitochondria play in HF with its manifold roles is presented in Fig. 18.1.
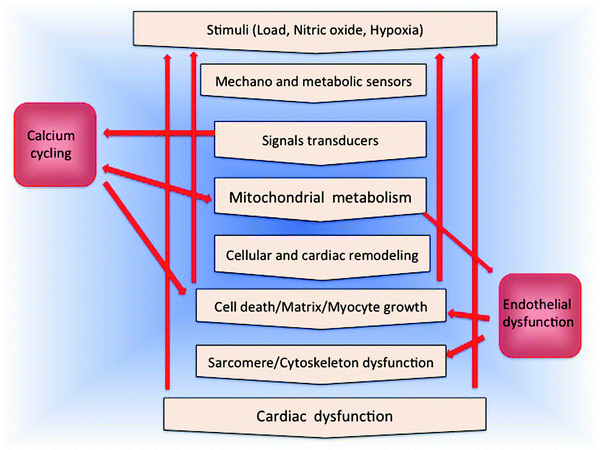
Fig. 18.1
Mitochondrial centrality in heart failure
In this chapter, a discussion on how mitochondrial dysfunction may be related to other critical cellular and molecular changes found in HF, including dysfunctional structural and cytoskeletal proteins, apoptosis, calcium flux and handling, and signaling pathways will be presented. Moreover, mitochondrial biochemical and molecular changes occurring in severe HF secondary to primary cardiomyopathy (dilated/hypertrophic) in humans as well as in animal models of HF secondary to volume and/or pressure overload will be examined. Finally, the available evidence on the role that mitochondrial dysfunction plays in HF will be presented.
Mitochondrial Bioenergetics in HF
Observations from animal models of HF, as well as from clinical studies, suggest that mitochondrial bioenergetic defects may underlie several aspects of the HF phenotypes. Besides their contribution to bioenergetic function, mitochondria are integrally involved in the regulation of intracellular Ca2+ flux, myocyte cell death, and remodeling events; in reactive oxygen species (ROS) generation and antioxidant response; and in furnishing cardioprotective responses to physiological insults.
The organelle manifold roles in bioenergetic production, sensor, and transport, as well as its effect on ROS generation and signaling, cell death, Ca2+ levels, and contractility, have often been overlooked (Table 18.1). In this section, we will focus on the mitochondrial changes that occur in HF, mainly on mitochondrial bioenergetics and biogenesis, ROS and oxidative stress (OS), and downstream effects on myocyte function (i.e., contractility and stimuli response) and cell death (i.e., apoptosis and necrosis). This discussion requires a larger view of mitochondria within the cellular context including interactions and cross talk with other organelles in the development and progression of HF. Current approaches to understand the role(s) that the organelle plays in the failing heart should include observations from experimental animal models, use of transgenic models, gene profiling analysis, in vitro studies with isolated cardiomyocytes (to identify pathway components in HF), and identification of potential targets for therapeutic interventions.
Table 18.1
Mitochondrial changes in heart failure
Alterations in the electron transport chain (ETC) and in oxidative phosphorylation (OXPHOS) |
Decreased respirasome-like supercomplex |
Defective respirasome organization, probably related to changes in the mitochondrial inner-membrane phospholipid composition or to modifications of the subunits of the ETC complexes |
Decreased levels of tricarboxylic acid cycle (TCA) |
Decreased levels of fatty acid oxidation (FAO) |
Increased production of reactive oxygen species (ROS) and oxidative stress (OS) |
Increased ROS-mediated damage to DNA, proteins, and lipids |
Changes in mitochondrial biogenesis and turnover |
Abnormal mitochondrial energy transfer via the adenine nucleotide translocator (ANT) and mitochondrial creatine kinase |
Abnormal membrane potential |
HF secondary to calcium concentration changes in the perfused canine heart is accompanied by a severe reduction in mitochondrial respiratory function |
Abnormal mitochondrial permeability transition |
Many of the major apoptotic proteins located in the mitochondria may impact on mitochondrial function and signaling |
ATP Generation
The heart is highly dependent for its function on oxidative energy generated in mitochondria, primarily by fatty acid β-oxidation (FAO), electron transport chain (ETC), and oxidative phosphorylation (OXPHOS). Abundant in energy-demanding cardiac tissue, mitochondria constitute over one-third of the cardiomyocyte cellular volume (i.e., a greater proportion than found in skeletal muscle). Energy production in mitochondria depends on genetic factors which modulate normal mitochondrial function, including enzyme activity and cofactor availability, and on environmental factors such as the availability of fuels (e.g., sugars, fats, and proteins) and oxygen. In the postnatal and adult heart, fatty acids are the primary energy substrate for heart muscle ATP generation by OXPHOS and the mitochondrial respiratory chain, the most important supply of cardiac energy, whereas the fetal heart derives energy primarily from the oxidation of glucose and lactate supplied by the glycolytic pathway. The phosphotransferase enzymes, creatine kinase and, to a lesser extent, adenylate kinase, play a pivotal role in the distribution of ATP from its site of synthesis in the mitochondria to spatially distinct sites of ATP utilization within the cytosol [1].
Figure 18.1 depicts the manifold roles that mitochondria play in HF. Adequate fuel supply and in particular ATP levels are critical for myocardial contractility and electrophysiology [2–6]. ATP produced by OXPHOS is preferentially used to support myocyte contractile activity [7]. Indeed, mitochondria appear to be clustered at sites of high ATP demand and are organized into highly ordered elongated bundles, regularly spaced between rows of myofilaments (intermyofibrillar mitochondria, IMFM), and in contact with the sarcolemma (subsarcolemmal mitochondria, SSM) (see Fig. 3.1 in Chap. 3). Moreover, structural contacts between the sarcoplasmic reticulum (SR) and mitochondria have been demonstrated by electron microscopy with evidence of coordination between these organelles at the level of Ca2+ homeostasis and regulation of ATP production [8].
Fatty acids are the primary energy substrate for heart muscle ATP generation by mitochondrial OXPHOS and the ETC, the most important energetic pathway which provides over 90% of cardiac energy. On the other hand, the supply of ATP from other sources such as cytosolic glycolytic metabolism is limited in normal cardiac tissue. Mitochondrial-localized FAO spiral and the oxidation of carbohydrates, through the matrix-localized tricarboxylic acid (TCA) cycle (see Chap. 5), generate the majority of intramitochondrial NADH and FADH2, which are the direct source of electrons for the ETC and also produce a portion of the ATP supply (Fig. 18.2).
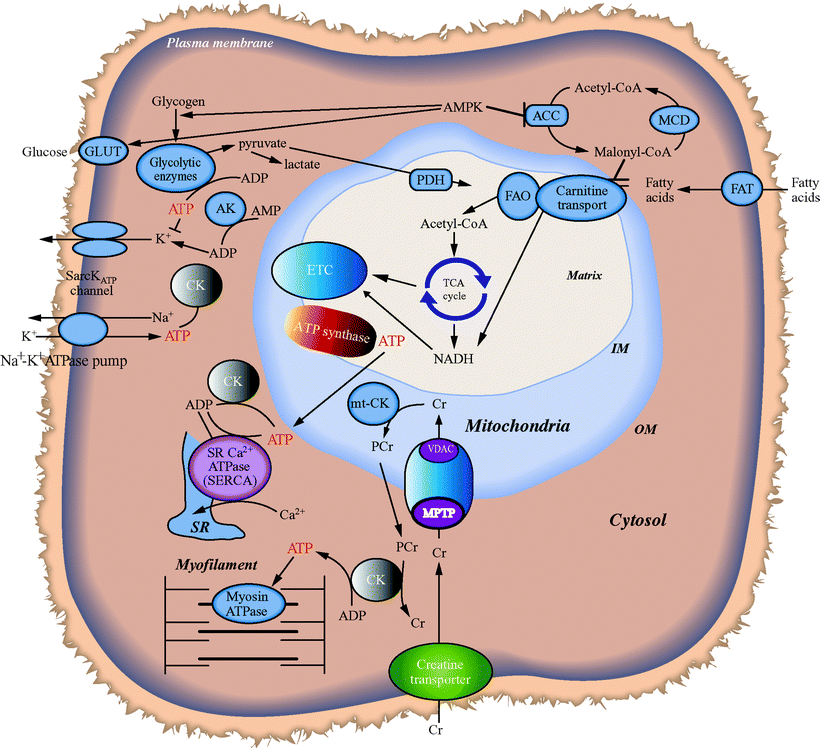
Fig. 18.2
Interplay of mitochondrial bioenergetic pathways with myocardial ATP utilization and FAO. MPTP mitochondrial permeability transition pore, VDAC voltage-dependent anion channel
The heart maintains stored pools of high-energy phosphates including ATP and phosphocreatine (PCr). The enzyme creatine kinase (CK), which has both mitochondrial and cytosolic isoforms, transfers the phosphoryl group between ATP and PCr at a rate estimated to be ten times greater than the rate of ATP synthesis by OXPHOS [9]. Under conditions that increase ATP demand in excess of ATP supply such as in acute pump failure in ischemia, utilization of PCr via the CK reaction is an important mechanism that maintains steady myocardial ATP levels. Another relevant enzyme-mediated transfer system dedicated to maintaining ATP levels includes adenylate kinase (AK) transferring phosphoryl groups. In addition, the adenine nucleotide translocators (ANT), a family of inner-membrane proteins that exchange mitochondrial ATP for cytosolic ADP, provide new ADP to the mitochondria while delivering ATP to the cytoplasm for cellular work support.
The concept of an energy-starved myocardium is supported by findings using powerful analytical technologies such as nuclear magnetic resonance (NMR) spectroscopy and positron emission tomography (PET) [2–6]. This concept has significant clinical implications in the management of patients with HF since pharmacological interventions that reduce metabolic demand, such as ACE inhibitors, angiotensin blockers, and β-blockers, improve clinical outcomes, while agents that increase metabolic demand, such as positive inotropic drugs, are less effective to ameliorate outcome and often they increase mortality [3, 4].
Reduced activity levels of mitochondrial bioenergetic enzymes, including selected respiratory enzymes and mitochondrial CK (mtCK), have been found in HF patients [10–13]. The failing human heart has a 25–30% decline in ATP levels as gauged from observations in heart biopsy specimens [14] and by 31p NMR spectroscopy [15]. Also, in animal models of HF, the loss of myocardial ATP is associated with a loss in the total adenine nucleotide pool, which is slow and progressive, suggesting that a decline in ATP content might only be detectable in advanced HF [16].
The total pool of cardiac creatine, which is phosphorylated by CK to form PCr, is significantly reduced in animal models of HF [17], and the extent of creatine depletion appears to correlate with the severity of HF in human [18]. Similarly, PCr levels as measured by [31]P NMR and PCr/ATP ratios were significantly lower in subjects with dilated cardiomyopathy (DCM) and HF [19]. Even at moderate workloads, a decrease in PCr/ATP ratio has been consistently reported in the failing human heart and in experimental HF. This is a strong predictor of cardiovascular mortality in patients with DCM (even better than left ventricle [LV] ejection fraction) [20]. Decreased amount of Cr transporter on the sarcolemma may decrease Cr accumulation in the failing myocardium [17, 21, 22]. The passage of Cr transporter to the plasma membrane is regulated by stress, insulin, growth factors, and mTOR, although little is known about the regulation of Cr transporter in the heart [23]. Potential mechanisms responsible for the decline in PCr may include reduced number or activity of the Cr transporter [24] and also alterations in CK expression/function [25]. While PCr levels or PCr/ATP is not a specific marker of HF (since they also decline in compensated LV hypertrophy), they are markers of mismatch between ATP supply and ATP demand for utilization.
There is clinical evidence that mitochondrial bioenergetic metabolism may be critical and primary in the development of HF. This information has been obtained from individuals with a variety of cardiomyopathies (CMs) secondary to specific nuclear gene defects in mitochondrial/metabolic proteins including ANT, respiratory complex enzyme subunits, molecules involved in complex IV assembly, and FAO enzymes (Table 18.2). Specific mitochondrial DNA (mtDNA) gene defects associated with clinical CM/HF (Table 18.3), often associated with neuropathy, have also been identified in tRNA genes [26], although mutations in structural genes such as ATP6/ATP8 and cytb (mitochondrial genes encoding subunits of complexes V and III, respectively) can also lead to CM/HF [27–29]. Henceforth, mitochondrial bioenergetic pathways including ETC, OXPHOS, TCA cycle, and FAO are crucial for the myocardial intracellular ATP-requiring pumps that control sarcomeric contractile functioning, calcium cycling, and transmembrane ion transport. While current evidence suggests that multiple deficits in these pathways likely contribute to bioenergetic decline in human HF, which is indicative of a programmatic shift in bioenergetic production and utilization, it is unclear when and how this occurs.
Table 18.2
Specific nuclear gene defects in mitochondrial metabolism leading to cardiomyopathy and HF
Gene loci | Protein function | Cardiac phenotype |
|---|---|---|
Nuclear genes | ||
PRKAG2 | AMP-activated protein kinase (regulatory subunit), energy sensor | HCM |
SCO2 | Cytochrome c oxidase (COX) assembly | HCM |
NDUFV2 | Respiratory complex I subunit | HCM |
NDUFS2 | Respiratory complex I subunit | HCM |
ANT | Adenine nucleotide transporter/mtDNA maintenance | HCM |
ACADVL | VLCAD activity (FAO) | HCM/DCM |
FRDA | Mitochondrial iron import | HCM/FA |
COX10 | Cytochrome c oxidase (COX) assembly | HCM |
SLC22A4 | Carnitine transporter (OCTN2) | HCM/DCM |
COX15 | Cytochrome c oxidase (COX) assembly | HCM |
TAZ (G4.5) | Tafazzin (FAO) | DCM/Barth syndrome |
MTP/HADHA | Mitochondrial trifunctional protein (FAO) | DCM |
Table 18.3
Specific mitochondrial gene defects leading to clinical cardiomyopathy and HF
Gene loci | Protein function | Cardiac phenotype |
|---|---|---|
MtDNA | ||
Leu3243 (A→G) | Mitochondrial tRNA biogenesis | DCM |
Leu3303 (C→T) | Mitochondrial tRNA biogenesis | Fatal CM |
Ile 4300 (A→G) | Mitochondrial tRNA biogenesis | HCM—adult onset, HF |
Ile 4320 (C→T) | Mitochondrial tRNA biogenesis | Fatal CM |
Ile4269 (A→G) | Mitochondrial tRNA biogenesis | CM, HF |
Lys 8334 (A→G) | Mitochondrial tRNA biogenesis | HCM, MERRF |
Lys 8363 (G→A) | Mitochondrial tRNA biogenesis | HCM, deafness, LS |
Lys 8296 (A→G) | Mitochondrial tRNA biogenesis | Fatal HCM |
Gly 9997 (T→C) | Mitochondrial tRNA biogenesis | Dysrhythmias, HCM |
16 S rRNA 3093 (C→G) | Mitochondrial tRNA biogenesis | MELAS, CM |
12 S rRNA 1555 (A→G) | Mitochondrial tRNA biogenesis | CM, HF |
Cytb 15498 (G→A) Gly→Asp | Cytochrome b, ETC | HiCM |
ATPase6 8993 (T→G) Leu→Arg | Mitochondrial ATP synthase, OXPHOS | LS, HCM |
Mitochondrial ROS Generation and Antioxidant Response
One attractive hypothesis on the contribution that mitochondria have in HF is related to its role in ROS production. As discussed in Chap. 10, the majority of intracellular ROS, including superoxide and hydroxyl radicals and hydrogen peroxide (H2O2), are a by-product of normal mitochondrial metabolism and bioenergetic activities (Fig. 18.3).
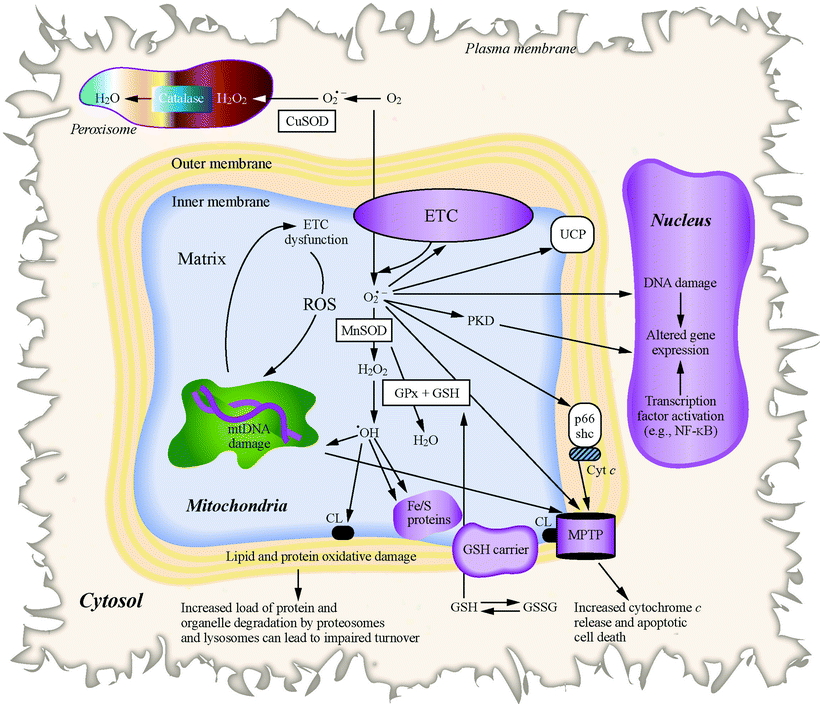
Fig. 18.3
Events leading to mitochondrial ROS and oxidative stress. Changes in proteins, lipids, and mtDNA caused by mitochondrial-generated ROS (including H2O2, superoxide [O2•−], and hydroxyl radicals (•OH)) lead to MPTP opening and apoptotic cell death, mitochondrial enzyme dysfunction, and nuclear DNA damage, as well as modulated DNA repair and gene expression in the nucleus. Antioxidant factors in peroxisomes (catalase), mitochondria (GPx, GSH, and MnSOD), and cytosol (CuSOD), which are scavengers of ROS, are also shown
Side reactions of mitochondrial respiratory enzymes (primarily complexes I and III) with oxygen directly generate superoxide anion radical; either excessive or diminished electron flux at these sites can stimulate the autoxidation of flavins and quinones (including coenzyme Q) producing superoxide radicals. The superoxide radicals can react with NO to form peroxynitrite, which is a highly reactive and deleterious free radical species, or can be converted by superoxide dismutase (SOD) to H2O2 that can further react to form highly reactive hydroxyl radicals. The high reactivity of the hydroxyl radical and its extremely short half-life of 10−9 s restrict an area of its damaging efficiency to a small radius from its origin since it is too short-lived to diffuse at a considerable distance. ROS generated by mitochondria can produce extensive oxidative damage to macromolecules such as proteins, DNA, and lipids, particularly targeting proximal mitochondrial components including mitochondrial respiratory enzymes, matrix enzymes (e.g., aconitase), and membrane phospholipids, mainly cardiolipin. Mitochondrial ROS-caused damage also affects a wide spectrum of cardiomyocyte functions such as contractility, ion transport, and calcium cycling. ROS generated in mitochondrial organelle also play a role in cell signaling (e.g., in triggering cardioprotective pathways) and in the transcriptional activation of specific nuclear genes eliciting a novel transcriptional programming.
In clinical cases, a clear link between OS/ROS and chronic ventricular dysfunction has only been established in anthracycline-mediated and alcoholic cardiomyopathies. In contrast, it remains unclear whether ROS or OS has a pathophysiologic role in the vast majority of patients with congestive HF or CM due to ischemic, hypertensive, valvular, or idiopathic causes [30]. Superoxide anions as assessed by electron paramagnetic resonance (EPR) spectroscopy with an O2•− spin trapping were reported to increase more than twofold in the failing ventricular myocardium from patients with end-stage HF undergoing transplant [31]. Moreover, despite increased mitochondrial-localized SOD (MnSOD) mRNA levels, a marked decline in MnSOD protein and activity was detected. Both increased ROS levels and decreased antioxidant response would be expected to lead to enhanced OS in the failing heart, which in turn may result in increased transcription of antioxidant enzymes. That excessive ROS/OS in HF may serve as a potent trigger for changes in specific nuclear gene expression and may also underlie the programmatic shift in mitochondrial bioenergetic function previously discussed, as well as acting as a catalyst in myocyte remodeling.
Critical Role of Mitochondrial Bioenergetic Enzymes and ROS in Animal Models of HF
That cardiac metabolism plays a central role in HF has been confirmed by O’Brien et al. observations of significant decrease in ATP-utilizing enzymes including SR Ca2+-ATPase (SERCA) and myofibrillar Ca2+-ATPase activities in dogs paced to advanced HF (4 weeks of pacing), whereas in dogs with early HF (1 week of pacing), there was a significant decline in activities of the SR Ca2+ release channel, mitochondrial ATP synthesis, and Cr [32, 33].
Data obtained from biochemical analysis in paced dogs documented a significant decrease in cardiac respiratory complex III and V activities [34]. Included in this study was a limited temporal course analysis that showed significant reduction in specific ETC enzyme activities in early HF (between 1 and 2 weeks of pacing) [35]. Moreover, this early ETC dysfunction correlated with the appearance of markers of myocyte apoptosis and OS, whose presence has been previously described in paced failing hearts [36, 37]. Using electron spin resonance (ESR) spectroscopy to directly assess ROS levels in paced dog hearts, Ide et al. demonstrated a significant increase in superoxide anion levels in myocardial submitochondrial fractions; the elevated ROS production was attributed to the functional block of electron transport, resulting from a marked decrease in mitochondrial respiratory complex activities [38]. Furthermore, later studies by Ide et al. [39] using the paced dog HF model and modified ESR analysis showed a significant increase in hydroxyl radical levels and a significant positive correlation between myocardial ROS levels and increases in LV contractile dysfunction.
Several models of mechanically overloaded heart have presented further evidence of defective mitochondrial bioenergetics in HF. In both volume- and pressure-overloaded rat hearts [40], depletion of PCr occurs with LV hypertrophy. Patients with HF have shown marked reductions in levels of phosphocreatine (similarly decreased in HCM and DCM) [41], and reduced mCK activity and content were also demonstrated in a pressure-overloaded rat model [42]. In volume-overloaded hearts, alterations in myocardial high-energy phosphate (i.e., significantly lower myocardial PCr/ATP ratios) have been postulated to contribute to contractile/pump dysfunction occurring during exercise [43]. Interestingly, these mitochondrial abnormalities in function persisted despite adequate tissue oxygenation [44] and suggest that altered regulation of mitochondrial OXPHOS results in reduced PCr levels present in HF [45]. Modulation of OXPHOS activities could be achieved by differential substrate utilization, changes in ADP, inorganic phosphate, intramitochondrial NADH, and oxygen levels [46].
Data from several laboratories have shown that two major subpopulations of mitochondria are present in skeletal and cardiac muscles. One of these organelle subpopulations is associated with the sarcolemma (i.e., SSM), and the second population, the IMFM, is proximal to the myofilaments. A preferential decline in IMFM oxidative function in aged rats has been reported, with concomitant reduction of complex III and activities [47–49]. Presently, there is limited data concerning the different susceptibility of these mitochondrial subpopulations and its contribution to the overall mitochondrial dysfunction observed in HF. Analysis of skeletal muscle has shown a pacing-associated decline in complex I and III activities but limited to SSM mitochondria. On the other hand, a significant decline in complex V activity was detected in both IMFM and SSM (with a higher decline in the IMFM fraction). Taken together, these findings reveal that both discrete defects in bioenergetics and the onset of increased apoptosis are present in pacing-induced CM/HF and operative in the early stages of pacing.
The specificity and reproducibility of these changes suggest that these are not stochastic defects. Both changes were in parallel with elevated ROS/OS levels. The abnormal activity of several mitochondrial enzymes and the increased apoptogenic pathway appear to be underlain, at least in part, by an orchestrated shift in expression (both nuclear and mtDNA) of several respiratory chain subunits (e.g., cytb, ATP-β), mitochondrial bioenergetic enzymes (e.g., mtCK), global transcription factor (e.g., PGC-1), and apoptotic proteins (e.g., p53, p21).
Furthermore, these data also suggest that differences in specific mitochondrial subpopulations associated with distinct cardiac compartments may contribute to the observed enzymatic dysfunction observed in HF. Further studies appear to be necessary to unravel the genesis of the orchestration of the intercommunication involved and identify potential targets for new therapies.
Recently, Ashrafian et al. [50] have identified a mouse mutant, called Python, which develops DCM and HF secondary to a dominant fully penetrant mutation in the dynamin-1-like (Dnm1l) gene, which has been shown to be critical for mitochondrial fission. Heterozygous Python fibroblasts showed abnormal mitochondria and peroxisomes with reduced levels of mitochondrial enzyme complexes and cardiac ATP depletion. Of interest, the Python mutation affected mitochondrial function prior to major changes in heart structure and before there was any overt signs of HF. The homozygous Python mutation resulted in the death of embryos midway through gestation. As the authors indicated, this is the first time that a defect in a gene involved in mitochondrial remodeling can result in CM, implying that the function of this gene is necessary to maintain normal cellular function in a relatively tissue-specific manner.
In another model of HF, the murine model of myocardial infarct (MI) created by ligation of the left anterior descending coronary artery for 4 weeks, Ide et al. found that LV dilation and contractility dysfunction were accompanied by significant increases in levels of mitochondrial ROS (i.e., hydroxyl radicals) and lipid peroxides [51, 52]. Moreover, infarcted LV from MI mice exhibited diminished enzymatic activity of respiratory complexes I, III, and IV (enzymes each containing subunits encoded by mtDNA), while the mitochondrial enzymes encoded only by nuclear DNA (e.g., citrate synthase and complex II) were unaffected. These investigators also reported a parallel decrease in steady-state levels of several mtDNA-encoded transcripts, including subunits of complexes I, III, and IV as well as 12S and 16S rRNAs and a significant decline in mtDNA copy number. These data are consistent with the concept that membrane lipids, mtDNA, and mitochondrial respiratory enzymes are well-established targets of ROS oxidative damage (see Fig. 18.3), albeit the absence of effect on complex II, a particular common ROS target, was unexpected.
Another marker of enhanced OS in the mouse failing heart was increased level of mitochondrial 8-oxo-dGTPase, a DNA repair enzyme which prevents the incorporation of 8-oxo-dGTP into DNA, a significant type of ROS-induced DNA damage which can lead to defects in DNA replication. This HF model illustrates both the development of mitochondrial ROS and a number of its downstream deleterious effects (e.g., enzyme dysfunction, mtDNA damage) as well as an adaptive response (i.e., increased DNA repair activity) to remove that damage.
It is also important to note that while a number of animal studies have shown ROS activation in the failing heart, both arising from mitochondrial and extramitochondrial sources, some observations have suggested that endogenous antioxidants and ROS defense pathways can ameliorate ROS-mediated cardiac abnormalities, and the dynamic balance between these counteracting forces should be considered.
Since direct measurement of short-lived ROS is extremely difficult, several laboratories have reported increased levels of ROS-mediated damage (e.g., lipid peroxidation, DNA and protein oxidation) as an indirect index of ROS/OS in several animal models, including in the pacing-induced HF [34, 36, 53]. The LV from paced animals exhibited increased aldehyde levels and marked reductions in the activity of respiratory complexes III and V together with increased levels of large-scale mtDNA deletions [34].
While elevated ROS activation in the failing heart has been shown to arise from both mitochondrial and extramitochondrial sources, the role of endogenous antioxidants in ameliorating myocardial OS and the dynamic balance between these counteracting forces should be considered. It is well known that both cytosolic antioxidant enzymes (e.g., catalase, SOD1/CuSOD) and mitochondrial-localized antioxidants including SOD2/MnSOD, thioredoxin, and glutathione peroxidase can reduce ROS levels. Also, thioredoxin and thioredoxin reductase form an enzymatic antioxidant and redox regulatory system implicated in the regeneration of many antioxidant molecules, including ubiquinone, selenium-containing substances, lipoic acid, and ascorbic acid [54].
Both cytosolic antioxidant enzymes (e.g., catalase, SOD1, thioredoxin) and mitochondrial-localized antioxidants including SOD2, thioredoxin, and glutathione peroxidase can reduce ROS levels. Moreover, thioredoxin and thioredoxin reductase form an enzymatic antioxidant and redox regulatory system implicated in the regeneration of many antioxidant molecules, including ubiquinone (Q10), selenium-containing substances, lipoic acid, and ascorbic acid [54]. As we will see later, the relationship of antioxidants with HF has been underlined by several transgenic mouse studies.
While the mechanism by which the antioxidant response signaled in the presence of ROS remains unknown, recent observations suggest that mitochondria can act as an OS sensor and signal to the nucleus to upregulate antioxidant production. Storz et al. have shown that mitochondrial ROS release can activate a signal relay pathway in which the serine/threonine protein kinase D (PKD), acting as a mitochondrial sensor of OS, activates the NF-κB transcription factor leading to induction of SOD2 [55, 56]. PKD has been localized to the mitochondria in cells exposed to both exogenous and mitochondrial ROS, where it is phosphorylated and activated. Upon subsequent dissociation from mitochondria, activated PKD phosphorylate substrates participate in NF-κB activation, although how this is accomplished is not yet known. Experiments in which RNAi-mediated silencing of PKD blunted SOD2 induction confirm that SOD2 promoter activation is dependent on PKD. While the role of PKD in HF has not yet been established, the critical involvement of mitochondrial ROS in the signaling and generation of an antioxidant response suggests potential limitations with the use of antioxidants, which target mitochondrial ROS.
Although studies with animal models suggested that ROS and defective ETC play an integral role in the genesis and progression of HF, these findings were correlative and did not, however, establish a direct link between ROS, defective ETC, and either skeletal muscle or cardiac dysfunction. Notwithstanding, these observations did foreshadow recent findings using heart-/muscle-specific MnSOD-deficient mice, a transgenic mice in which OS, which selectively impaired mitochondrial respiration, can directly cause HF [57]. Furthermore, ROS may trigger a multiplicity of mechanisms that can lead to HF. For instance, complex IV (affected at both the transcriptional and activity levels in Ide’s studies) was not significantly decreased in the MnSOD-deficient mouse. On the other hand, this model exhibited a moderate decline in both complexes I and III and, particularly, a marked decline in the nuclear DNA-encoded complex II activity (shown to be mediated at the posttranscriptional level). Taken together, this latter study and the aforementioned pacing dog studies do not support the view of ROS-mediated decline in mtDNA copy number and in mitochondrial transcription as the definitive mechanism leading to reduced respiration and cardiac bioenergetic metabolism in HF.
Finally, studies using a porcine model of LV infarction and remodeling (induced by ligation of the left circumflex artery), reduced systolic performance accompanied by reductions in high-energy phosphate levels, myocardial OXPHOS, and concomitant reductions in ANT protein and in the β subunit of complex V were noted [58, 59]. These findings suggest that specific mitochondrial inner-membrane proteins may play a critical role during the remodeling that occurs in HF. Nevertheless, the order of the myocardial molecular and biochemical events leading to abnormal regulation of OXPHOS needs to be established.
Electron Transport Chain Respirasome in HF
The concept that the mitochondrial ETC is integrated by an orderly array of individual enzymatic complexes spreading freely in the inner membrane has been challenged by the discovery of a supramolecular organization of these complexes. Schagger and Pfeiffer [60] reported that all cytochrome c oxidase (complex IV) of Saccharomyces cerevisiae is bound to cytochrome c reductase (complex III), which exists in three forms: the free dimer and two supercomplexes comprising an additional one or two complex IV monomers. The distribution between these forms varies with the growth conditions. In mammalian mitochondria, almost all complex I was assembled into supercomplexes comprising complexes I and III and up to four copies of complex IV, a model for a network of respiratory chain complexes termed “respirasome.” Also, a fraction of total bovine ATP synthase (complex V) was isolated in a dimeric form, suggesting that a dimeric state is not limited to S. cerevisiae but also exists in mammalian mitochondria.
Observations by Shagger et al. [61] on the formation of respirasomes in human revealed that the mitochondrial ETC is organized in supramolecular assemblies; complexes I (NADH dehydrogenase) and III (cytochrome c reductase) formed a stable core respirasome to which complex IV (cytochrome c oxidase) may also bind. Analysis of the state of respirasomes in patients with an isolated deficiency of single complexes revealed that the formation of respirasomes is essential for the assembly/stability of complex I, the principal entry point of respiratory chain substrates. Moreover, genetic defects leading to a loss of complex III prevented respirasome formation and led to the secondary loss of complex I. Thus, primary complex III assembly deficiencies may manifest as a combined complex III/I defects.
The detection of supercomplexes and their constituent complexes became possible by using 2-D native electrophoresis, a technique pioneered by Schagger and von Jagow [62] employing the net negatively charged dye Coomassie Brilliant Blue G-250, which allows/improves the migration of proteins in a gel matrix driven by an electric field. Moreover, digitonin and blue native electrophoresis (BN-PAGE) have been applied for the initial separation of supercomplexes, followed by less mild native electrophoresis variants in the second dimension to release the individual complexes from the supercomplexes. By reducing the complexity of these supramolecular systems to the level of individual complexes, 2-D native systems may allow not only the identification of constituent proteins, but also their copy number [62]. In addition, the involvement of complex IV in the supramolecular organization of the ETC in photosynthetically active tissues of higher plant mitochondria has been possible in digitonin extracts of partially purified mitochondria of green plants using BN-PAGE and a gentler colorless native (CN)-PAGE; in this model a substantial proportion of the respiratory complex IV was assembled with complexes I and III into respirasome-like supercomplexes [63]. It is worth noting that in the case of supramolecular structure separation, in particular the ATP synthase, the Coomassie Brilliant Blue G-250 may need to be omitted. As pointed out by Seelert and Dencher [64], even for monomeric ATP synthase, this dye leads to partial dissociation into the subcomplexes F1 and Fo, and only ATP synthases of some species are unaffected by this treatment. Thus, for applications with fragile complexes, clear or colorless native PAGE (CN-PAGE) and/or the variant high-resolution CN-PAGE are the methods of choice (as depicted in Fig. 18.4).
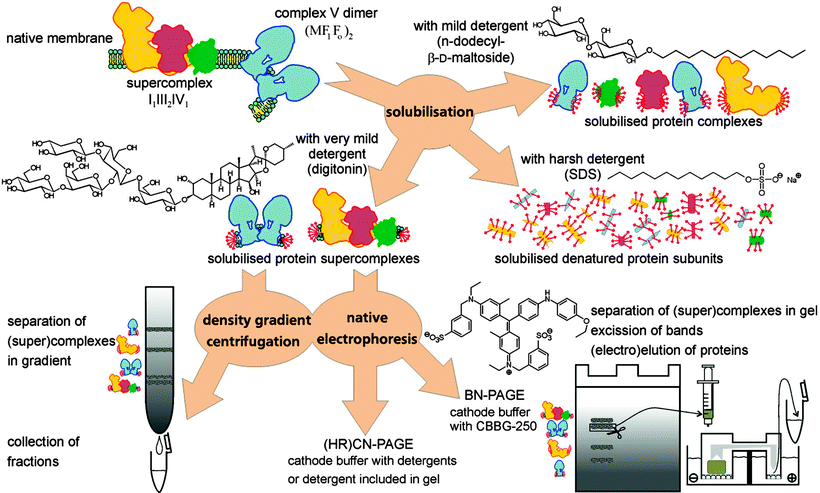
Fig. 18.4
Isolation of supramolecular structures of ATP synthase and respiratory chain supercomplexes. Density gradient centrifugation and preparative native electrophoresis are frequently employed as purification steps prior to solubilization. Samples from density gradients can be directly employed for functional and structural studies. Native gels are applicable for in-gel activity tests, but in-depth study requires elution of the supercomplexes from the gel matrix (adapted from Seelert & Dancher [64] with permission from Elsevier)
The first 3-D map of an ETC supercomplex has been recently reported by Schafer et al. [65] using random conical tilt electron microscopy analysis of a bovine supercomplex consisting of complex I, dimeric complex III, and complex IV (I1III2IV1). In this 3-D map, the location and orientations of all the individual complexes in the supercomplex were distinctively achieved as seen in Fig. 18.5
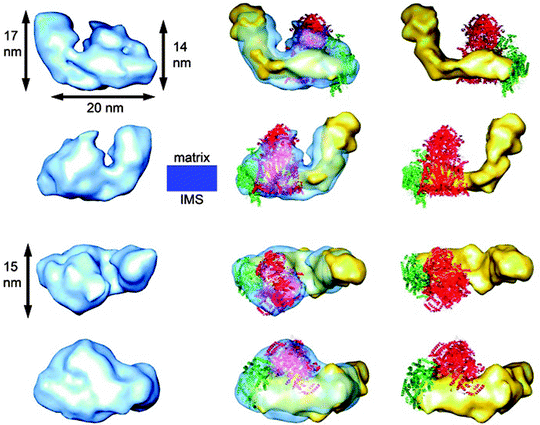
Fig. 18.5
Comparison of the 3-D map of supercomplex I1III2IV1 with those of the individual complexes. The left column shows a surface representation in blue of the supercomplex I1III2IV1 3-D map. The middle column displays the 3-D map with a semitransparent surface and the fitted structures of the individual complexes. The right column shows only the three individual complexes as they would assemble to form the supercomplex. The two upper rows display side views along the membrane plane. The third row shows the particles as seen from the matrix space and the lower row as seen from the intermembrane space. The electron microscopic 3-D map of complex I is shown in yellow, the X-ray structure of complex III2 in red, and the X-ray structure of complex IV in green. The location of the membrane in a side view is displayed in blue. IMS = intermembrane space (reprinted with permission from Schafer et al. [65] Copyright 2011. American Chemical Society)
Moreover, the ubiquinone and cytochrome c binding sites of each complex in the supercomplex were also located as depicted in Fig. 18.6.
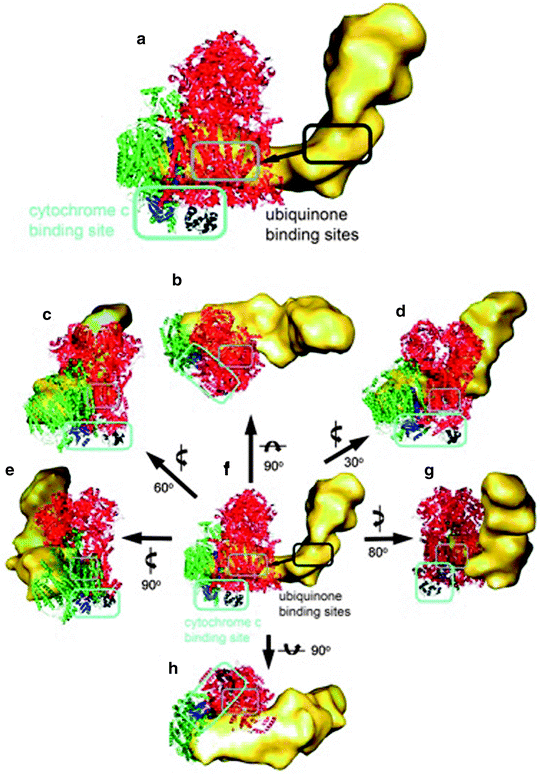
Fig. 18.6
Electron carrier binding sites in the supercomplex. (a) Magnified side view along the membrane plane. (b) Top view from the matrix side. (c−g) Side views along the membrane plane. (h) Bottom view from the intermembrane space. The individual complexes in the supercomplex are shown in yellow (complex I), red (complex III2), and green (complex IV). (a, f) The ubiquinone interaction site in complex I is marked in dark gray, and the interaction site in the complex III dimer is marked in gray. In all other representations only the ubiquinone interaction site in the complex III dimer is marked in gray. The two cytochrome b L hemes of the complex III dimer, which are the ubiquinone interaction site, are shown in light gray. The ubiquinone diffuses from its interaction site in complex I to its interaction site in the complex III dimer, shown as a dark gray arrow in (a) and (f). The cytochrome c interaction site in complexes III and IV is marked in light blue in (a−h). Cytochrome c bound to complex III is shown in black, and subunit II, which is the cytochrome c binding site, from complex IV is displayed in blue. The rotations to obtain the different views are shown next to the arrows (reprinted with permission from Schafer et al. [65] Copyright 2011. American Chemical Society)
The mobile electron carrier binding site of each complex was found to be in proximity to the binding site of the succeeding complex in the respiratory chain allowing structural evidence for direct substrate channeling in the supercomplex assembly with short diffusion distances for the mobile electron carriers. Also recently, Dudkina et al. [66] employing cryoelectron tomography and subvolume averaging reported the 3-D reconstruction of bovine heart respirasome (Fig. 18.7).
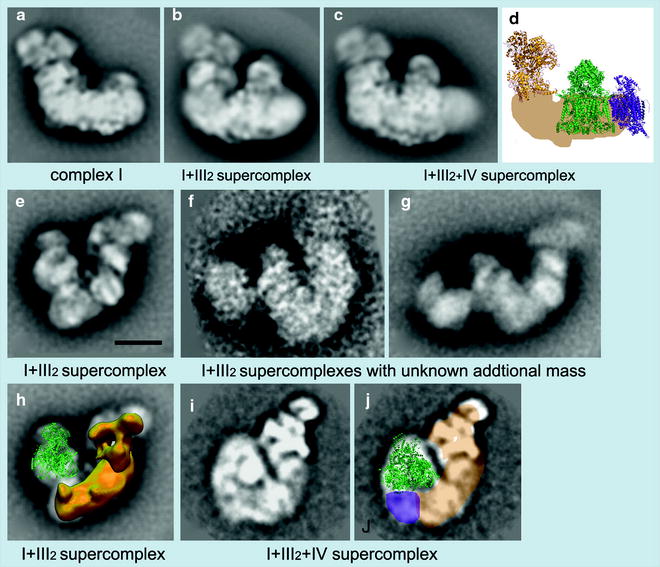
Fig. 18.7
Examples of EM projection maps of OXPHOS supercomplexes obtained by negative staining (a, b, c, e, f, g, h) and cryo-EM (i, j) plus modeling. (a–c) Averaged side views of bovine complex I and its supercomplexes. (d) Positions of individual complexes within the I + III2+IV supercomplex or respirasome from bovine mitochondria [67]. Yellow X-ray structure of the hydrophilic domain of complex I from Thermus thermophilus [68], green dimeric bovine cytochrome c reductase (complex III [69], purple bovine cytochrome c oxidase (complex IV) [70]. (e) Top view of the I + III2 supercomplex from maize [71]. (f, g) Top views of I + III2 supercomplexes with an unknown additional mass from bovine and potato mitochondria, respectively (Dudkina et al., unpublished data). (h) Assignment of the complexes I and III2 within the top-view map of I + III2 supercomplex from maize, as presented in image (e); the complex III dimer is in green in a tilted position; in yellow the 3-D EM structure of complex I from Yarrowia [72]. (i) Map of the bovine respirasome (N.V. Dudkina, unpublished data). (j) Modeling of the respirasome. In beige the position of the membrane arm of complex I, in green X-ray structure of the bovine dimeric complex III, and in purple the position of a complex IV monomer. Scale bar, 10 nm (reprinted from Dudkina et al. [66] with permission from Elsevier)
It was composed of dimeric complex III and single copies of complexes I and IV, at near 2.2-nm resolution. Unexpectedly, the distance between cytochrome c binding sites of complexes III2 and IV was about 10 nm, and modeling indicated a loose interaction between the three complexes, which suggest that lipids are gluing them at the interfaces. These investigators have also observed a stable, dimeric ATP synthase supercomplex using mitochondria of the alga Polytomella. Structural analysis by electron microscopy and single-particle analysis revealed that dimer formation is based on specific interaction of the Fo parts, not the F1 headpieces which are not at all in close proximity. The angle between the two Fo parts was about 70°, which allowed marked local bending of the membrane. It seems that the function of ATP synthase dimerization is to control the particular architecture of the mitochondrial inner membrane [73].
There is evidence that the assembly of the functional respirasome is altered in HF. Rosca and Happle [74] have proposed a sequential mechanistic pathway in which the decrease in functional respirasome in HF is the primary event causing decreased OXPHOS and increased ROS production, leading to a progressive decrease in cardiac performance. Moreover, using native gel electrophoresis in the canine model of coronary microembolization HF, they found a dramatic decrease in ADP-stimulated respiration that was not relieved by an uncoupler, although the ADP/O ratio was normal, which indicates no defect in the phosphorylation apparatus. These findings inferred the presence of a defect in OXPHOS within the ETC, while the individual ETC enzyme activities were within normal limits. The respirasome-like supercomplex (complex I/complex III dimer and at least a significant proportion of complex IV) was decreased indicating that the mitochondrial defect lied in the supermolecular assembly rather than in the individual components of the ETC [75].
Furthermore, whether a decrease in the respirasome organization was due to changes in the phospholipid composition of the mitochondrial inner membrane or to modifications of the subunits of the ETC complexes has been analyzed by Rosca et al. [76]. The contents of the main phospholipid species, including cardiolipin, as well as the molecular species of cardiolipin were found unchanged in the failing heart mitochondria. However, employing phosphatase inhibitors, a specific decrease in complex I-supported respiration with glutamate was found. On the other hand, employing saponin-permeabilized cardiac fibers, preincubation with cAMP decreased OXPHOS, likely due to a localized defect at complex IV of the ETC. Taken together, these findings suggest that phosphorylation of specific complex IV subunits decreases OXPHOS either by limiting the incorporation of complex IV in supercomplexes or by decreasing supercomplex stability. Moreover, assembly of the ETC into respirasomes can provide structural support for mitochondrial OXPHOS, by facilitating electron channeling and perhaps by preventing electron leak and superoxide production. As Garcia-Palmer [77] has pointed out, these newly identified ETC assembly mechanisms open a new area of research into mitochondrial functional changes in general and in HF in particular. Further research on the structural conformation, presence, and assembling of mitochondrial supercomplexes will improve our understanding of their mechanistic implications and also increase the potential to develop new diagnostic methods and treatments for mitochondria-related diseases.
Transgenic Models in the Study of Mitochondria Alterations in Heart Failure
In mice transgenic models, disruption of mitochondrial bioenergy at specific loci or pathways can cause CM and HF. Gene ablation in mice (i.e., the generation of null mutations or gene knockouts) targeting a relatively wide spectrum of genes encoding specific mitochondrial proteins results in severe CM. Notably, mutational inactivation of the heart/muscle isoform of Ant1 gene in transgenic mice will result in the development of skeletal myopathy and CM leading to HF [78]. The Ant1 gene-deficient mice exhibit mitochondrial abnormalities including a partial deficit in ADP-stimulated respiration, consistent with impaired translocation of ADP into mitochondria in both skeletal muscle and heart. Ant1 − / − mice also exhibit a progressive cardiac hypertrophic phenotype coincident with the proliferation of mitochondria [79]. This mitochondrial biogenic response may be a compensatory mechanism to correct the energy deficit but could also be contributory to cardiac remodeling. Null mutations in either the mitochondrial or cytosolic CK gene in mice also lead to increased LV dilation and hypertrophy [42, 80]. Palmieri et al. [81] have reported the presence of a recessive mutation in the heart-/muscle-specific isoform of ANT1 in a patient with HCM and mild myopathy with exercise intolerance and lactic acidosis. This mutation resulted in complete loss of adenine nucleotide transport function and was also associated with increased levels of muscle mtDNA deletions.
Besides ANT [79], other targeted genes include MnSOD [82], glutathione peroxidase (GPx) [83], and factors involved in fatty acid metabolism, e.g., mitochondrial permeability transition pore (MPTP) subunits [84], mitochondrial transcription factor A (TFAM) [85], and frataxin [the protein responsible for Friedreich’s ataxia (FRDA)] [86]. Interestingly, ANT-deficient mice develop progressive HCM while MnSOD-deficient mice develop DCM, yet both types of null mutation cause severe cardiac ATP deficiency which is thought to underlie the resulting cardiac phenotype(s). Another major contribution of the transgenic mouse model has been to further our understanding of the family of transcriptional coactivators and factors including PPAR-α, PGC-1α, NRF-1, and MEF-2, which coordinately regulate myocardial energy metabolism, and their essential role in the developing embryonic heart, as well as to delineate the order of biochemical and molecular events in the metabolic and transcriptional cascade governing energy regulation in both the normal and diseased hearts [87].
In addition to examining the specific effects on cardiac phenotype by eliminating specific nuclear genes regulating mitochondrial function, tissue-specific knockout mice with mitochondrial CM have been used to identify modifying genes of potential therapeutic value [88]. At present, there is limited information about the impact on myocardium of knocking out nuclear genes involved directly in mitochondrial OXPHOS. A recent report described cardiac dysfunction in mice lacking cytochrome c oxidase subunit VIa-H, the heart isoform [89]. In addition, the “knockout” approach has not yet been accomplished with mtDNA genes due to the formidable technical difficulty involved in direct gene replacement or ablation of a multicopy gene in the setting of a nonnuclear multicopy organelle (i.e., the mitochondrion), although several promising approaches will be discussed in a later chapter.
The use of cardiac-specific overexpression of specific genes has also proved highly informative in furthering our understanding of the role of mitochondria in cardiac dysfunction. This technique involves fusing a regulatory region from a cardiac-specific gene with a candidate gene of interest and introduction into the transgenic mice, which will express the candidate gene specifically in cardiac muscle cells. Overexpression of genes which mediate the expression and control cardiac energy metabolism (e.g., PGC-1α, PPAR-α) has been shown to lead to severe cardiac dysfunction and marked changes in mitochondrial structure and function [90–92]. Similarly, transgenic mice containing cardiac-specific overexpression of calcineurin exhibited severe cardiac hypertrophy (which progresses to HF), marked mitochondrial respiratory dysfunction, and superoxide generation [93].
The development of animal models of mitochondrial-based cardiac dysfunction offers the possibility of direct testing for potential treatments. For example, the demonstration that MnSOD-deficient animals developed ROS toxicity and DCM prompted speculation that effective treatment with antioxidants could ameliorate the cardiac phenotype; indeed, peritoneal injection of MnSOD-deficient mice with the antioxidant MnTBAP [manganese (III) tetrakis (5,10,15,20 benzoic acid) porphyrin] eliminated the cardiac dysfunction and reversed the ROS accumulation [94]. Overexpression of the antioxidant GPx in transgenic mice inhibited the development of LV remodeling and failure after myocardial infarct (MI) and was associated with the attenuation of myocyte hypertrophy, apoptosis, and interstitial fibrosis [95]. Moreover, Schriner et al. have demonstrated that overexpression of catalase (primarily a peroxisomal-localized enzyme) targeted to the mitochondria increased overall mouse longevity, diminished OS- and ROS-mediated mitochondrial protein and mtDNA damage, and delayed the onset of aging-mediated cardiac pathology including subendocardial interstitial fibrosis, vacuolization of cytoplasm, variable myofiber size, hypercellularity, collapse of sarcomeres, mineralization, and arteriosclerosis, changes commonly observed in elderly human hearts and often found in association with congestive HF [96]. Another important finding was that it mattered in which subcellular compartment the overexpressed catalase was localized with little evidence of benefits from nuclear or peroxisomal-localized as compared to striking benefits of mitochondrial-localized catalase activity.
Among the most compelling evidences supporting a primary role for mitochondrial ROS and OS in CM and HF are findings with antioxidant genes in transgenic mice. Strains harboring null mutations in either MnSOD or TrxR2 encoding mitochondrial thioredoxin reductase exhibit DCM and HF. [57, 82, 97, 98] Li et al. [82] found that mice homozygous for MnSOD deficiency resulted in early neonatal death from severe DCM and metabolic acidosis. Moreover, these strains displayed severe reduction in myocardial succinate dehydrogenase (complex II) and aconitase (a TCA cycle enzyme) suggesting that MnSOD is required for maintaining the integrity of mitochondrial enzymes susceptible to direct inactivation by superoxide.
Mice, in which MnSOD deficiency was targeted to skeletal muscle and heart (i.e., H/M-Sod2 −/− strains), displayed progressive HF with depressed cardiac contractility by 8 weeks, cardiac enlargement by 16 weeks, and death from HF by 22 weeks [82]. Cardiac pathology was associated with specific defects in mitochondrial respiration (i.e., severely reduced respiratory complex II and moderately reduced complex I and III activities) and in myocardial mitochondrial ultrastructure. Immunoblot analyses showed significant expression of the SDHA and SDHB subunits from myocardial complex II of H/M-Sod2 −/− mice, with moderate suppression of complex Iα 9, Rieske iron–sulfur protein and core I subunit of complex III, and α and β subunits of complex V. Mitochondrial superoxide production was also significantly higher in these mice as was mitochondrial (but not cytosolic) lipid peroxidation, suggesting that oxidative damage was specifically localized in mitochondria. In addition, myocardial ATP production and content were significantly diminished, which may account for the absence of energy-dependent apoptosis in H/M-Sod2 −/− mice. This study also offered further evidence that ROS and OS are intimately linked to the progression of CM/HF since the administration at 8 weeks of age of the antioxidant MnSOD mimetic (MnTBAP) significantly improved cardiac contractility and ameliorated the overall phenotype.
It has been suggested that OS and alterations in protein metabolism may play an essential role in the pathogenesis of FRDA [99]. Schulz et al. [100] have assessed whether induction of mitochondrial metabolism may prevent the development of CM and HF. Transgenic mice were generated overexpressing the mitochondrial protein frataxin, which promotes mitochondrial energy conversion by controlling iron–sulfur cluster biogenesis and hereby mitochondrial electron flux. In this model, following doxorubicin-induced experimental CM, cardiac function and survival were significantly improved. Since the cardiac insulin/insulin-like growth factor 1 (IGF-1) cascade is markedly inhibited in wild-type mice following induction of CM, it seems that transgenic overexpression of frataxin may rescue the impaired insulin/IGF-1 signaling, providing enhanced cardiac stress resistance in the transgenic mice. These observations suggest that increased mitochondrial metabolism elicits an adaptive response due to mildly increased OS as a consequence of increased oxidative energy conversion; this in turn activates protective mechanisms which counteract cardiotoxic stress and promote survival in experimental CM.
Mitochondrial DNA and HF in Transgenic Mice
ANT1, TFAM, and DNA polymerase γ are among the many metabolic-related genes for which either deletion or overexpression can promote the development of CM and HF in transgenic mice. While TFAM and DNA polymerase γ play well-established roles in the maintenance, replication, and transcription of mtDNA, ANT which is pivotally involved in mitochondrial ATP/ADP exchange and transport, as well as a component of the MPTP, has been reported to have a role in mtDNA maintenance, possibly arising from its participation in the regulation of deoxynucleotide levels [101].
As noted before, mutational inactivation of the mouse Ant1 gene encoding the heart/muscle isoform of the mitochondrial ANT results in mitochondrial abnormalities including a partial deficit in ADP-stimulated respiration, consistent with impaired translocation of ADP into mitochondria in both skeletal muscle and heart. In addition, Ant1 deficient mice exhibit multiple myocardial mtDNA deletions associated with elevated production of ROS (e.g., H2O2) and the development of skeletal myopathy and CM leading to HF [78]. Moreover, Ant1-deficient mice displayed an increase in tissue-specific antioxidant defenses (e.g., MnSOD) in skeletal muscle mitochondria but not in heart mitochondria. Ant1 −/− mice exhibit a progressive cardiac hypertrophic phenotype coincident with the proliferation of mitochondria [79]. It has been suggested that this mitochondrial biogenic response is a compensatory mechanism to correct the energy deficit, although could also be contributory to cardiac remodeling.
Transgenic mice heterozygous for a null allele of TFAM showed decreased myocardial mtDNA copy number and ETC defects, whereas homozygous TFAM knockout strains exhibited severe mtDNA depletion with decreased OXPHOS function and died in embryonic development [102]. Wang et al. [85] reported that mouse strains containing conditional cardiac- and muscle-specific null TFAM alleles developed a mosaic pattern of progressive and severe ETC defects in the postnatal heart, resulting in DCM, atrioventricular conduction block, early HF, and death between 2 and 4 weeks. Reduced activities of complexes I and IV together with a significant decline in cardiac and skeletal muscle mtDNA levels and gene expression were reported. Along with the development of severe CM, tissue-specific TFAM knockout mice exhibited increased apoptosis in the in vivo heart consistent with the finding of massive apoptosis in Tfam knockout embryos and suggesting that defects in ETC may predispose cells to apoptosis [103]. Global gene profiling analyses in tissue-specific Tfam knockout mice revealed a metabolic switch in the early progression of cardiac mitochondrial dysfunction akin to the activation of a fetal gene expression program in which a number of genes, encoding critical enzymes in FAO, showed decreased expression, while several genes encoding glycolytic enzymes showed increased expression [104]. In more advanced disease, the metabolic switch was followed by an increase in mitochondrial biomass or biogenesis which did not result in increase overall myocardial mitochondrial ATP production rate. It was inferred, on the basis of these findings, that the observed switch in metabolism appeared unlikely to benefit energy homeostasis in the respiratory chain-deficient hearts and may actually promote further cardiac dysfunction.
TFAM overexpression in transgenic mice can ameliorate the mitochondrial dysfunction and HF resulting from myocardial infarction [105]. TFAM overexpression attenuated the decline in mtDNA level and respiratory activities in post-MI hearts and significantly reduced the LV dilatation and dysfunction accompanied by a decrease in LV remodeling (i.e., decreased myocyte hypertrophy and interstitial fibrosis). The survival of the infarcted animals was affected, but not the infarct size.
Cumulative damage to mtDNA, which can result in point mutations, large-scale deletions, or changes in mtDNA copy number, has been implicated in the progression of HF. Mice that express a proofreading-deficient mitochondrial DNA polymerase γ targeted to the heart generated cardiac mtDNA mutations (average 2 per mitochondrial genome) and eventually (over several weeks) developed DCM and interstitial fibrosis, often leading to HF [106]. Surprisingly, the mechanism of the pathogenesis in these strains does not appear to involve increased OS levels [107]. Measurements of enzyme function or oxidative defense systems in the transgenic heart fail to detect increased levels of oxidative adducts in DNA or protein nor signs of increased OS. Furthermore, mitochondrial respiratory function and mitochondrial ultrastructure and number remained normal in these strains although the detection of cytochrome c release from mitochondria, a landmark of apoptosis, suggested that the elevated frequency of mtDNA mutations might trigger the initiation of apoptotic cell death. Interestingly, further studies have noted that the activation of myocardial programmed cell death pathway precedes (and may itself trigger) a vigorous prosurvival response including the upregulation of antiapoptotic proteins Bcl-2, Bcl-xl, Bfl1, heat shock protein 27, and X-linked inhibitor of apoptosis protein (XIAP) [108].
Peroxisome Proliferator-Activated Receptor-γ Coactivator 1 (PGC-1α)
The heart appears to function best when it simultaneously oxidizes both carbohydrates and fatty acids as bioenergetic substrates [109]. Under pathologic conditions, the heart relies more on glucose, as seen in cardiac hypertrophy, or may rely almost solely on fatty acids, as observed in cardiac tissue of animal models of diabetes [110]. Furthermore, the failing heart exhibits a decline in overall mitochondrial oxidative catabolism (including FAO), which is most evident in advanced HF while reliance on anaerobic glycolytic pathways tends to be increased [111–113]. Recent observations in an animal model of HF have downplayed the role of myocardial FAO downregulation in early, compensated HF [114]. Thus, initially this switch in metabolic substrate provides adequate energy to maintain normal cardiac function and may have an adaptive function by diminishing oxygen consumption. However, over time substrate switch in concert with declining OXPHOS becomes maladaptive, leading to a state of myocyte energy insufficiency (related to reduced capacity for myocardial mitochondrial ATP production and depletion in high-energy phosphates), resulting in diastolic HF. As previously noted, alterations in high-energy phosphates (PCr and ATP) have been identified by magnetic resonance spectroscopy both in animal models and human hearts with LV hypertrophy or HF [113, 115] and may be contributory to the pathological remodeling that occurs in end-stage HF.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


