(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Abnormalities in cardiac mitochondrial respiratory enzymes and mitochondrial DNA have been found in an increasing number of pediatric cases of both dilated and hypertrophic cardiomyopathy, giving rise to the entity known as mitochondrial cardiomyopathy. Also, the association of mitochondria defects, including mitochondrial DNA mutations, with other pediatric congenital and acquired cardiac diseases is increasingly recognized. Histochemical, biochemical, and molecular findings in mitochondrial cardiomyopathy; mitochondrial defects associated with neurological disorders (“cardioneuropathies”); and mitochondrial alterations in congenital and acquired pediatric heart defects are reviewed in this chapter.
Introduction
While congenital heart disease (CHD), cardiomyopathy, dysrhythmias, and acquired cardiac diseases are common causes of mortality and morbidity in infants and children, the basic underlying mechanisms of many specific pediatric cardiovascular diseases still remain undetermined. Breakthroughs in molecular biology and genetic technology have just begun to be applied in pediatric cardiology stemming from the use of chromosomal mapping and the identification of genes involved in both the primary etiology and as significant risk factors in the development of cardiac and vascular abnormalities. For example, in DiGeorge syndrome, which often presents with CHD such as tetralogy of Fallot, the human mitochondrial citrate transporter gene (SLC20A3) maps to chromosome band 22q11, within a region implicated in DiGeorge syndrome, velocardiofacial syndrome, and schizophrenia. Also in Barth syndrome, an X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria, the locus has been mapped to Xq28. Interestingly, defects in the structure and function of mitochondria have also been found in an increasing number of children with CHD, including patent ductus arteriosus (PDA), right ventricular dysplasia, and dysrhythmias. Also, loss of myofibrils and accumulation of mitochondria, with infrequent formation of lipofuscin bodies characterizing degenerating muscle fibers, have been reported in CHD.
In this chapter the focus will be on information obtained thus far by biochemistry, molecular biology, and molecular genetic analysis in the diagnosis, treatment, and overall understanding of pediatric cardiovascular disease pathogenesis, examining both the bioenergetics and molecular abnormalities in cardiomyopathies and in the more prevalent congenital/inherited heart defects and sporadic and acquired disorders. Moreover, the association of cardiomyopathy with neurologic disorders, so-called cardioneuropathies, will be also reviewed.
General Aspects of Mitochondria
Since a detailed analysis of mitochondria structure and morphology has been presented in Section I of this volume, here it is suffice to note that the mitochondrial organelles are essential elements in energy generation and cell metabolism. Approximately 65% of mitochondrial proteins are localized in the matrix, 25% in the inner membrane, and the remaining proteins are in the outer membrane and intermembrane space. The electron transport system is localized in the inner membrane, and the energy generated is used to produce ATP via oxidative phosphorylation (OXPHOS). Mitochondrial energy production depends on both nuclear and mitochondrial DNA-encoded genetic factors which modulate normal mitochondrial function and on environmental factors including the availability of substrate fuels (e.g., sugars, fats, and proteins) and oxygen. Several interacting bioenergetic pathways contribute to mitochondrial energy metabolism including pyruvate oxidation, the tricarboxylic acid (TCA) cycle, the mitochondrial β-oxidation of fatty acids, and the common final pathway, OXPHOS (Fig. 12.1).
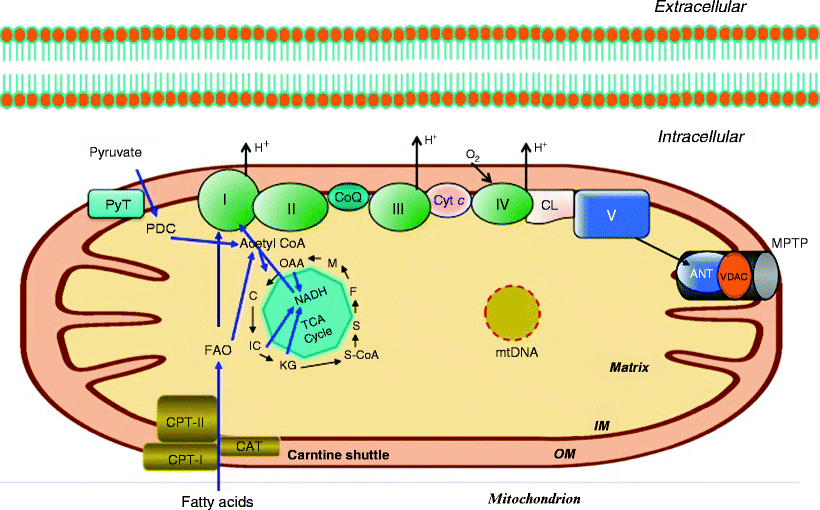
Fig. 12.1
Mitochondrial major metabolic reactions involved in OXPHOS. Respiratory complexes I–V are illustrated showing directional proton movement (H+) as indicated. The electron carriers CoQ (coenzyme Q) and Cyt c (cytochrome c) are shown. The TCA cycle is depicted with key metabolic intermediates including malate (M), oxaloacetic acid (OAA), citric acid (C), isocitrate (IC), α-ketoglutarate (KG), succinate (S), succinyl-CoA (S-CoA), fumarate (F), and NADH. Also depicted are the carnitine shuttle pathway for the mitochondrial import of fatty acids, including carnitine palmitoyltransferases (CPT-I and CPT-II) and carnitine-acylcarnitine translocase (CAT); the pyruvate transporter (PyT); pyruvate dehydrogenase complex (PDC); adenine nucleotide translocator (ANT); mitochondrial permeability transition pore (MPTP); and voltage-dependent anion channel (VDAC). Also, the general organization of the mitochondrial compartments is shown. Other abbreviations: CL mitochondrial IM phospholipid cardiolipin; IM mitochondrial inner membrane; mtDNA mitochondrial DNA; OM mitochondrial outer membrane
OXPHOS is performed by complexes of proteins located at the mitochondrial inner membrane including the electron transport chain (ETC) complexes I–IV, ATP synthase (complex V), and the adenine nucleotide translocator (ANT). Fatty acids are the primary energy substrate for the production of ATP in cardiac muscle by OXPHOS. In order to be utilized for bioenergetic production via mitochondrial fatty acid β-oxidation, fatty acids need to be effectively transported into the cardiomyocyte and subsequently into the mitochondria, a process requiring several transport proteins (fatty acid translocase, carnitine acyltransferase and two carnitine palmitoyl transferases, as well as carnitine). Fatty acid β-oxidation and the oxidation of carbohydrates via the TCA cycle generate the majority of intramitochondrial NADH and FADH2 which are the direct source of electrons for the electron transport chain. The supply of ATP from other sources (e.g., glycolytic metabolism) is limited in normal cardiac tissue. In addition to these bioenergetic pathways and metabolic intermediates, the heart also maintains stored high-energy phosphates (e.g., ATP and phosphocreatine pools).
The human mitochondrial genome has its own double-stranded circular molecule of DNA (mtDNA) encompassing 16,569 bp and encoding 13 proteins which constitute a portion of the 5 enzyme complexes involved in the OXPHOS-respiratory chain. The protein-encoding mtDNA genes are transcribed into specific mRNAs (Fig. 12.2) which are translated on a mitochondrial specific ribosome/protein synthesis apparatus. The mtDNA also encodes part of the mitochondrial protein synthesis machinery including 2 rRNAs and 22 tRNAs.
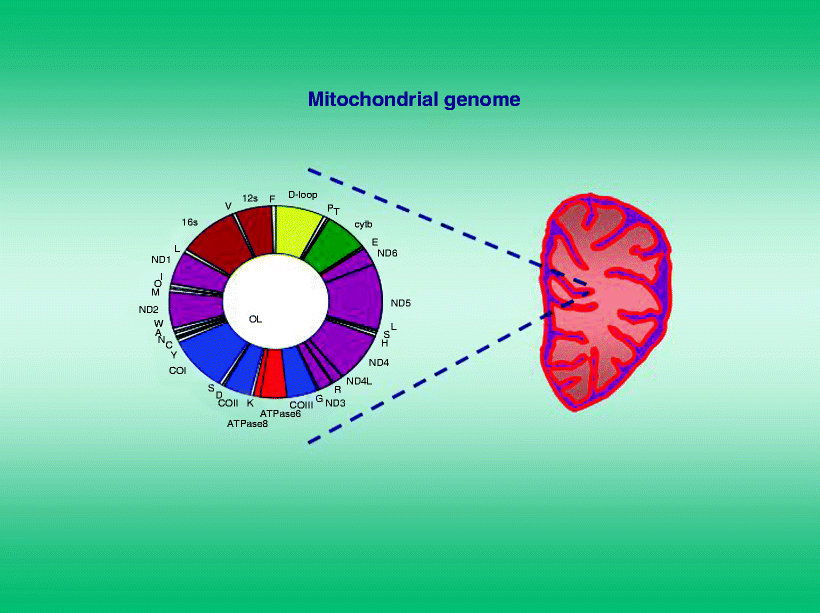
Fig. 12.2
The human mitochondrial genome
The biogenesis of myocardial mitochondria significantly increases during cardiac hypertrophy and also during treatment with thyroxin, electrical stimulation, and exercise; however, the mechanism(s) that regulates cardiac-specific mtDNA levels is not known nor is the mechanism of overall regulation of mitochondrial number.
As pointed out in Chap. 1, the singularity of the mitochondria organelle is having its own DNA, transcription, and translation systems. This mtDNA is strictly maternally inherited, does not recombine, and has a high incidence of spontaneous mutations. In mammalian cells, mtDNA is a small double-stranded circular molecule containing two ribosomal RNAs, 22 transfer RNAs, and 13 structural genes encoding some of the proteins of the five multisubunit enzyme complexes involved in OXPHOS. The remaining mitochondrial proteins in the multisubunit enzyme complexes—as well as the manifold proteins involved in mtDNA replication, mtRNA transcription, and their regulation—and the protein components of the mitochondrial ribosome, structural proteins, and transport proteins in the mitochondrial membranes are all encoded by the nucleus. Therefore, mutations in a large number of nuclear genes involved in mitochondrial biogenesis might be expected to result in cardiac enzyme and mtDNA defects, including an increased incidence of large-scale mtDNA deletions and mtDNA depletion associated with cardiac disorders. The nuclear genes encoding these mitochondrial proteins are inherited in a Mendelian manner, showing either autosomal or sex-linked inheritance. Thus the genetics of OXPHOS are rather complex since the enzymes involved are encoded by both the nuclear and the mitochondrial genomes.
Anomalies in mitochondrial structure and OXPHOS function result in OXPHOS diseases [1, 2] and appear to have particularly dramatic consequences for the energetics of the cardiac cell. Moreover, the majority of OXPHOS diseases identified thus far are caused by mtDNA mutations and are maternally inherited.
Mitochondrial Cardiomyopathy
Mitochondrial cardiomyopathy (MCM) can be defined as an OXPHOS disease characterized by abnormal cardiac mitochondria either in number, structure, or function. By altering the ETC function, specific pathogenic mtDNA mutations or depletion of mtDNA levels may also result in cardiomyopathy. MCM may present a hypertrophic phenotype, characterized primarily by left ventricular or biventricular hypertrophy and often accompanied by myofibril disarray, or a dilated phenotype characterized by increased ventricular size and impaired ventricular function. Moreover, cardiomyopathy with neurological disorders (“cardioneuropathies”) will be discussed in a latter subsection. Here it is suffice to say that MCMs may occur in association with neurological disorders such as MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), MERFF (myoclonic epilepsy and ragged red fibers), Kearns–Sayre syndrome (KSS), and Leigh syndrome (LS) [1, 2]. This association likely stems from the strong demand that both neural (e.g., brain and ocular tissue) and cardiac tissues have for oxidative energy as well as their high sensitivity to its deprivation. The above disorders may present early in childhood, whereas others may manifest themselves later (adult or adolescent onset). Moreover, mitochondrial defects have also been noted in a number of cases of fatal infantile cardiomyopathy [3–6].
Biochemical studies have demonstrated that specific defects in mitochondrial ETC and/or OXPHOS are the major loci of mitochondrial dysfunction in MCM [1, 7]. Since the five multisubunit respiratory complexes (I–V) located in the mitochondrial inner membrane consist of over 70 different types of polypeptide components, a large number of potential sites for defects are located in the pathway for energy generation (see Fig. 12.1).
In MCM as well as in other OXPHOS diseases, single or multiple defects in each of the respiratory complex activities might be present [4, 8–10]. Some of these defects can be secondary to changes in mtDNA, such as specific mutations in the mitochondrial protein synthesis apparatus, e.g., mitochondria-encoded tRNAs [4] or mutations in structural protein-encoding genes [11, 12] (Table 12.1); however, in the majority of MCMs, the primary site of defect has not yet been elucidated.
Table 12.1
Genetic defects in pediatric cardiomyopathy
Gene product affected (gene locus) | Cardiac phenotype | References |
|---|---|---|
Metabolism and bioenergetics | ||
Mitochondrial trifunctional protein (HADHA) | Dysrhythmias, DCM | |
Carnitine palmitoyltransferase II (CPT2) | Dysrhythmias, SCD, CM | |
Carnitine-acylcarnitine translocase (CACT/SLC25A20) | Dysrhythmias, SCD, CM | |
Carnitine transport (OCTN2/SLC22A5) | HCM, DCM | |
Tafazzin (TAZ/G4.5) | DCM (Barth) | [25] |
Mitochondrial Fe2+ metabolism-frataxin (FRDA) | HCM (Friedreich ataxia) | |
Very-long-chain acyl-CoA dehydrogenase (ACADVL) | HCM, SCD | [28] |
Lysosomal α-glucosidase, acid maltase/glycogen storage (GAA) | Ventricular preexcitation; HCM (Pompe’s disease) | |
Glycogen-debranching enzyme (AGL) | HCM (Cori’s disease) | |
α-galactosidase (GLA) | HCM (Fabry’s disease) | |
Mitochondrial heme metabolism (COX15) | Early-onset fatal HCM | [35] |
γ-2 subunit of AMP-activated protein kinase (PRKAG2) | HCM, conduction defects (WPW) | |
Mitochondrial DNA | ||
tRNAleu, tRNAlys, tRNAile, tRNAgly | HCM (MELAS, MERRF) | |
ATPase6 | HCM (Leigh) | [39] |
Sporadic mtDNA deletions | Conduction defects (KSS) | [40] |
The pattern of maternal inheritance has been used to corroborate the presence of a mitochondria-based pathology in a number of familial cardiomyopathies [4, 41, 42]. On the other hand, mutations in nuclear genes may also be associated with MCM. For instance, a mutation in a nuclear gene involved in mtDNA replication has been implicated in an autosomally inherited cardiomyopathy characterized by accumulation of multiple mtDNA deletions in the patient’s brain and heart [43, 44]. Mutations in two nuclear genes NDUFV2 and NDUFS2, encoding subunits of respiratory complex I, have been also implicated in the development of early-onset HCM [45, 46].
Initially, the term mitochondrial cardiomyopathy (MCM) was used [10] to describe the phenotype of a group of young patients (n = 16) with respiratory enzyme defect with either DCM or HCM. Four of these patients harbored specific pathogenic point mtDNA mutations likely involved in enzymatic and bioenergetic defects. The criteria that were used to evaluate the pathogenicity of mutations include being heteroplasmic (albeit in high proportion relative to the wild-type allele) and to be present in highly conserved residues/nucleotides. Mutations found in tRNAArg, cytb, and ND5 satisfy these rigorously defined criteria for pathogenic alleles. The nature of the enzymatic defects in patients harboring these pathogenic alleles is also consistent with their role in the etiology of the enzyme defects. For instance, complex III activity is markedly reduced in those cases harboring pathogenic cytb mutations. It is noteworthy that the mtDNA mutations reported in this study were not detected in other studies of mtDNA mutations in cardiomyopathy [47, 48]. Similarly, previously detected mtDNA mutations were not found in our patients with the exception of the cytb15236 mutation [49]. These results are further indication of the heterogeneity of mitochondrial DNA defects, making the job of routine screening for pathogenic mtDNA mutations particularly difficult since relatively few mutations are shared.
We share with other investigators the view that cardiomyopathy with ETC defects is more common than previously thought and tends to follow a different and more severe clinical course; however, the incidence of mitochondrial DNA mutations are usually found in a minority of patients, suggesting that most mitochondrial disorders of childhood follow a Mendelian pattern of inheritance [50]. Recent observations suggest that heart involvement is a common feature associated with early- and late-onset forms of ETC defects, and in particular cases, these conditions should be considered within the diagnostic algorithm of idiopathic cardiomyopathies [51].
Fatal infantile cardiomyopathy can result from the severe skeletal and cardiac muscle complex IV deficiency found in cases with mutations in the nuclear SCO2 gene. This gene encodes the copper-binding protein involved in the assembly of cytochrome c oxidase (COX) subunits into a functional complex IV enzyme [52].
MCM can also occur sporadically. Various agents that cause damage to the mitochondria can result in cardiomyopathy. Some of these agents may have nonspecific or multiple modes of action on both mitochondria and the cell, e.g., alcohol, adriamycin, and ischemia. The relationship of mtDNA damage to cardiomyopathy is of great interest since somatically generated mtDNA deletions (mainly 5-kb and also 7.4-kb deletions) as shown in Fig. 12.3 increase during myocardial ischemia [53]. In addition, KSS, a neuromuscular disorder with atrioventricular conduction defects and cardiomyopathy, is commonly associated with abundant large-scale mtDNA deletions as shown in Fig. 12.3 whose generation is thought to be spontaneous, since they are rarely detected in mothers or siblings.
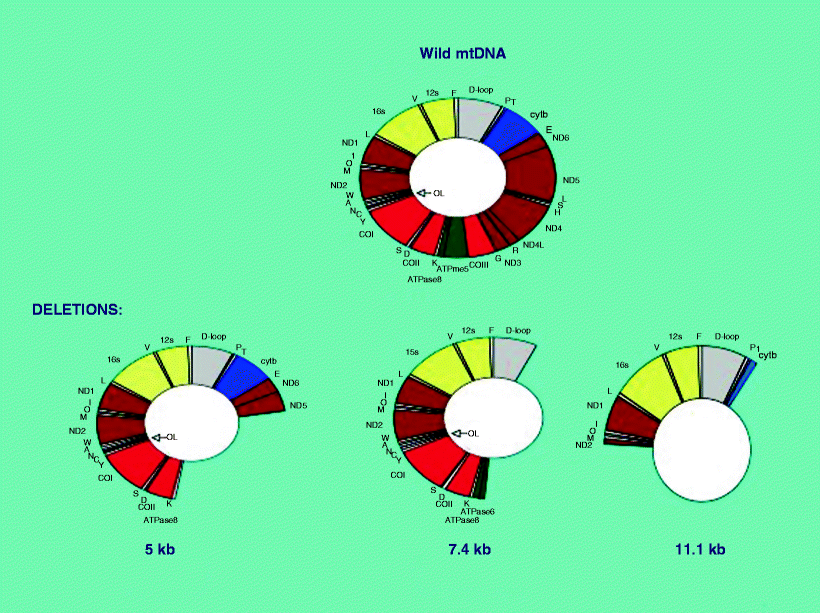
Fig. 12.3
Wild-type mtDNA and mtDNA deletions. The 11.1-kb deletion was detected in a case of KSS
In contrast, a severe cardiomyopathy associated with multiple mtDNA deletions and progressive external ophthalmoplegia (PEO) is transmitted by an autosomally recessive nuclear gene defect [54]. This defect along with mutations in other nuclear genes such as Twinkle, ANT1, and polymerase subunit g (POLG) presumably affects the proteins involved in mtDNA maintenance, leading to development of multiple mtDNA deletions with cardiac defects. This unique etiology of cardiac pathology, involving both Mendelian transmission and mitochondrial cytopathy, has been termed a “dual genome” disease [55, 56]. As previously noted, in most clinical cases, the molecular basis and the pathogenetic significance of mtDNA deletions in the cardiomyopathic heart remain undefined. Among the agents that have been shown to result in specific mitochondrial damage and cardiomyopathy are adriamycin and zidovudine (AZT) [57, 58]. Recently, using proteomics, Stěrba et al. [59] provided novel information on the effect of chronic anthracycline toxicity in the heart. Cardiotoxicity in rabbits (employing daunorubicin, a member of the anthracycline family, 3 mg/kg, weekly, 10 weeks) showed changes in the left ventricle proteome analyzed by two-dimensional difference gel electrophoresis (2D-DIGE). Defects were found in mitochondria, especially in proteins essential for OXPHOS, energy channeling, antioxidant defense, and mitochondrial stress. In addition, the intermediate filament desmin, which interacts with mitochondria, was upregulated and disorganized in its expression pattern. These latter changes reflect the intensity of toxic damage in the whole heart as well as in cells. Furthermore, a marked drop in myosin light chain isoforms, activation of proteolytic machinery (including the proteasome system), increased abundance of chaperones and proteins involved in chaperone-mediated autophagy, membrane repair, as well as apoptosis were present.
Cardioneuropathies and Mitochondrial Phenotype
Previously, we have defined cardioneuropathy (CN) as an entity characterized by cardiomyopathy, either dilated or hypertrophic, coexisting with neurologic disorders and variable mitochondrial phenotype. Some of these neurologic disorders may be primary and present with multisystemic involvement, including cardiac dysfunction arising from abnormalities in mitochondrial bioenergetic transduction. Several syndromes have been described, including MELAS and MERRF which appear to be caused by defined pathogenic mitochondrial DNA point mutations in specific mitochondrial transfer ribonucleic acids (tRNAs). These mutations affect mitochondrial DNA-encoded protein subunits of the respiratory chain complexes, which are pivotally involved in the generation of the cell’s bioenergy [1, 2]. Moreover, a group of multisystemic disorders with associated cardiomyopathy are attributable to mitochondrial defects originating in either mitochondrial DNA structural genes or nuclear DNA (e.g., Leigh disease, Friedreich’s ataxia [FRDA]). Other less well-characterized multisystemic disorders with cardiac involvement are more difficult to classify, albeit mitochondrial involvement appears to be frequently present.
To further understand the relationship between clinical and mitochondrial phenotypes in patients with this intriguing association, Marin-Garcia et al. [60] have reported a study of 28 young patients with both cardiomyopathy and neurologic disorders. These include seizures, dystonia, ophthalmoplegia, KSS, LD, and FRDA. All tissues (heart n = 15 and skeletal muscle n = 11) examined displayed marked defects in respiratory complex activities. Five patients had abundant large-scale mitochondrial DNA deletions, and one patient displayed a pathogenic point mutation previously reported with mitochondrial cytopathy. Patients with hypertrophic cardiomyopathy (HCM) (n = 14) displayed a higher incidence of complex I defects, fewer DNA deletions, and mitochondrial structural abnormalities and were less often associated with developmental delay phenotype compared with patients with dilated cardiomyopathy (DCM) (n = 14). Although structural abnormalities were present in a subset of patients, evaluation of respiratory enzyme activity was the most informative whether tissues examined were derived from heart or skeletal muscle (Table 12.2). One patient (16) had several signs indicative of KSS (i.e., ptosis, PEO, and atrioventricular conduction block), whereas a second case (13) had an atrioventricular conduction defect and hypotonia but no ptosis or PEO and died of cardiac failure at age 7 years. Two patients (1 and 2) shared both the neurologic signs and progressive cerebral deterioration symptomatic of Leigh disease. Two patients had a clinical presentation suggestive of FRDA (25 and 28). In the 25 patients for whom cardiac or skeletal muscle tissues were available, reduced levels of mitochondrial enzymatic activities were detected.
Table 12.2
Mitochondrial analysis: cardioneuropathy biochemical, molecular, and morphological data
Age/sex | Cardiac | DNA defects | Enzyme defects | Mitochondria structure | |
|---|---|---|---|---|---|
Cardiac tissue | |||||
1 | 3 years/M | HCM | 8993 | V | Abnormal |
2 | 5 months/F | HCM | None | V | Normal |
3 | 6 months/M | Severe HCM | None | I | Normal |
4 | 5 months/M | Severe HCM | 5- and 7.4-kb deletions | I, III | Increased mito number |
5 | 6 months/M | DCM | ND | I, V | NA |
6 | 3 months/M | Severe HCM | None | I, III, IV, V | NA |
7 | 2.5 years/M | Severe HCM neonatal-onset | None | III, V | Normal |
8 | 3 weeks/F | DCM | None | I, III, IV, V | NA |
9 | 17 days/M | Severe HCM | None | I | Abnormal |
10 | 3 months/F | DCM | None | I, V | NA |
11 | 20 years/M | Moderate DCM | 5-kb deletion | ND | Abnormal |
12 | 10 days/F | Mild HCM | ND | III | Increased lipid deposition |
13 | 7 years/F | Severe DCM/CAVB | None | I, III, IV | Abnormal |
Skeletal muscle | |||||
13 | 7 years/F | Severe DCM/CAVB | None | I, III, IV, V | Abnormal |
14 | 15 years/M | DCM/CABB | 5 kb and novel 11.1-kb deletions | I, III, IV, V | Abnormal RRF |
15 | 1 years/M | DCM | None | III, IV, V | Abnormal |
16 | 1 week/M | Mild DCM | None | III, V | NA |
17 | 2 months/F | Severe HCM | None | I, III, V | Normal |
18 | 9 months/F | Moderate DCM | ND | I, III, IV, V | NA |
19 | 8 years/M | Severe HCM | None | III, IV | Normal |
20 | 76 years/F | DCM | 5- and 7.4-kb deletions | I, III, V | Abnormal |
21 | 16 years/F | Moderate DCM | ND | IV | Abnormal |
22 | 10 months/F | DCM | None | I, IV, V | NA |
23 | 2 months/M | Severe DCM | Novel 4.4-kb deletion | I. III, V | Abnormal |
24 | 2.5 years/M | Mild DCM | Frataxin (both mutant and wild-type allele) | I, III | NA |
Blood | |||||
25 | 10 years/F | Severe DCM | 5- and 7.4-kb deletions | ND | Normal |
26 | 12 years/f | HCM | Frataxin (only mutant allele) | ND | NA |
The spectrum of mitochondrial respiratory enzymes defects is shown in Table 12.2.
Reduced enzyme activity levels were found in complex I (18 cases), III (18 cases), IV (9 cases), and V (16 cases). Complex II and citrate synthase activity levels were in all cases similar to controls. Reduced levels of multiple respiratory complex activities were noted in 20 cases, and reductions in single respiratory complex activities were found in 5 cases, 4 of which were detected in cardiac tissue. Patients with MCM had increased incidence of complex I deficiency (12/13) compared with cases with DCM (6/13).
Pathogenic mitochondrial DNA point mutations [4, 6, 12, 41, 61–68], which have been considered responsible for encephalomyopathies including MELAS, MERRF, LD, and HCM, were found in one patient (a mutation at 8,993 in a case with LD). Five patients had significant levels of large-scale mitochondrial DNA deletions, including the common 5- and 7.4-kb deletions as well as several novel mitochondrial DNA deletions; two patients exhibited single deletions, and three patients had multiple deletions. Four of the five patients with mitochondrial DNA deletions had DCM; 1 had HCM. The location of these deletions in the human mitochondrial genome is shown in Fig. 12.3. The patient (16) with the classic KSS phenotype displayed multiple mitochondrial DNA deletions, including a novel 11-kb deletion that substantially deleted cytb and ND2 and entirely deleted the structural genes COI, COII, COIII, ATP6, ATP8, ND3, ND4, ND4 L, and ND5, as well as 13 transfer RNAs, including tryptophan, alanine, asparagine, cysteine, tyrosine, aspartic acid, lysine, glycine, arginine, histidine, serine, leucine, and glutamine, as well as the origin of replication for the light strand, as shown in Fig 12.3. The younger patient with KSS (13) did not show mitochondrial DNA deletions; however, severe defects in multiple mitochondrial enzymes, present in both skeletal and cardiac muscle, suggest the potential involvement of an unscreened pathogenic transfer RNA mutation. Two patients with clinical presentation consistent with FA harbored abnormalities in the frataxin gene; one (28) had homozygous mutant alleles [with a high level of GAA repeats (>1,500)]; the second patient (25) appears to be heterozygous with a mutant allele containing a high level of GAA repeats (>1,700) as well as a wild-type-sized allele; the latter also had marked deficiency in complex I and III activity levels in skeletal muscle. Structurally abnormal mitochondria were demonstrated by electron microscopy in 8 patients (of 16 examined). Normal organelles were mixed with strikingly abnormal mitochondria with stacks of paracrystalline bodies with characteristic geometric internal structure (Fig 12.4).
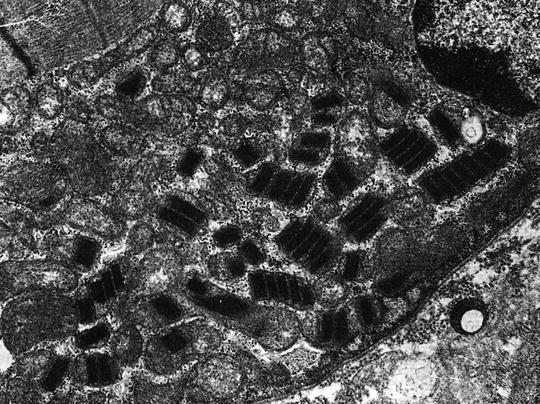
Fig. 12.4
Mitochondrial ultrastructure in a case of Kearns–Sayre syndrome (see text)
No evidence was found of myofibril disarray, nuclear or sarcolemmal abnormalities (e.g., typical of dystrophy), or inflammatory or necrotic lesions. Also, one patient displayed scattered ragged red fibers on light microscopy. One patient also displayed a marked increase in the number of cardiac mitochondria. Patients with HCM tended to have less evidence of mitochondrial structural abnormalities (3/9) compared with patients with dilated cardiomyopathy (5/7).
In summary, defects in mitochondrial DNA and bioenergetics are frequently present in children with cardiomyopathy presenting in association with neurologic abnormalities. The high incidence and severity of the mitochondrial alterations in both cardiac and skeletal muscles of very young patients with cardioneuropathy suggest that a mitochondrial cytopathy plays an important and potentially primary role in the pathogenesis of these disorders.
Diagnosis
Findings at the clinical, histochemical, biochemical, and molecular levels are employed to characterize MCM, keeping in mind that the highly variable clinical manifestations of MCM are indicative of both its heterogeneity and its frequent association with multisystemic disorders. Histochemical and morphological analysis to identify mitochondria morphology and number is helpful in some cases but is not always specific. Biochemical analysis demonstrating decreased specific respiratory enzymes activities is probably the most consistent assay for MCM.
However, biochemical analysis of the respiratory chain requires (in most cases) highly invasive endomyocardial biopsy, although enzyme analysis of biopsied skeletal muscle can also be informative [69]. Another important diagnostic tool in the study of MCM is the analysis of specific mtDNA alterations including point mutations and deletions, which are increasingly reported in cardiomyopathies. Molecular genetic analysis of mtDNA mutations in blood, biopsied skeletal muscle, or cultured cells can also be informative. A complete assessment of MCM should include both molecular, genetic, and biochemical analysis together with a detailed clinical profile.
Clinical and Laboratory Findings in MCM
MCM has often been found in association with certain “soft” clinical signs in patients and maternal relatives including hearing loss, hypotonia, and migraine-like headaches. Cardiac involvement includes both DCM and HCM, ventricular dysrhythmias, and conduction defects [70, 71]. Lactic acidosis is commonly present, and many patients display generalized muscle weakness [71, 72].
In a retrospective analysis of 90 young patients with mitochondrial myopathy, the incidence of cardiomyopathy was found to be near 6% and the prevalence of dysrhythmias around 22. Four patients received permanent pacemaker because of Adams–Stokes attack or bradydysrhythmias (mitochondriopathy diagnosis was made 1–3 years post pacemaker implantation in three cases). History of syncope, respiratory failure, right bundle branch block (RBBB), atrial fibrillation, and episodic ventricular tachydysrhythmias was presented in one patient with mitochondriopathy; another patient developed atrial tachydysrhythmias. Dysrhythmias were present in 14 patients including RBBB, bifascicular block, intraventricular block, Wolff–Parkinson–White (WPW) syndrome, and short PR interval syndrome. Eight patients exhibited a mtDNA 3243A-G mutation [73].
Histological and Electron Microscopic (EM) Analysis
Reported abnormalities in mitochondrial structure include distended cristae, electron-dense and paracrystalline inclusions, as well as swollen or distended mitochondrial membranes Fig. 12.4 and Fig. 12.5.
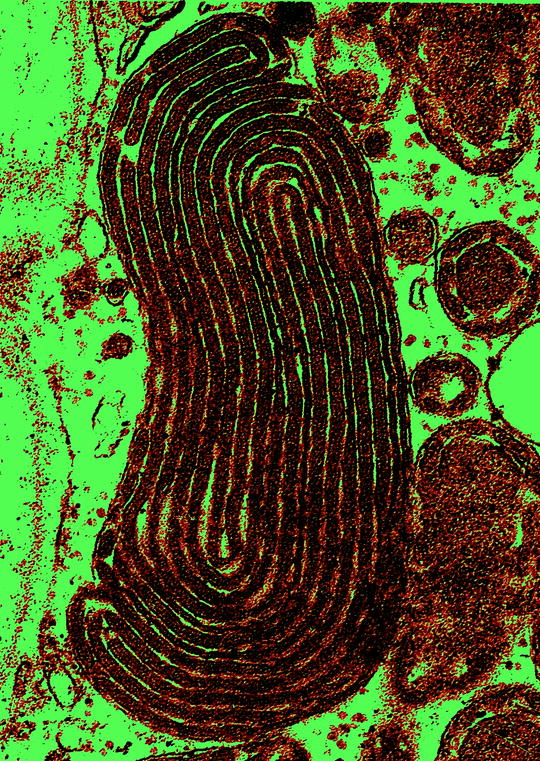
Fig. 12.5
Megamitochondria with lamellar-type crystalline inclusions
In skeletal muscle, ragged red fibers (RRFs) are often present (Fig. 12.6) and may be associated with disorders of adult/adolescent onset [74], mainly in disorders caused by defects in mitochondrial protein synthesis and rarely in disorders associated with mitochondrial structural gene defects [75].
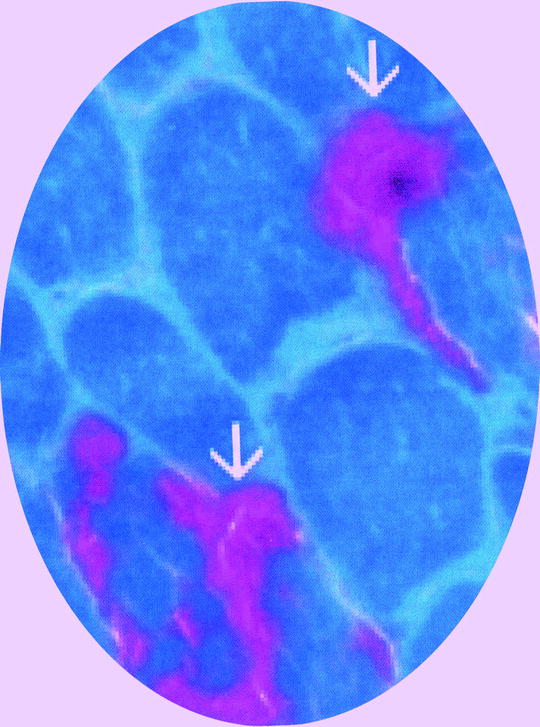
Fig. 12.6
Ragged red fibers (arrows) identified with modified Gomori trichrome stain. Normal myofibers are green
In addition, electron microscopy may allow the assessment of mitochondria number and structural abnormalities (Fig. 12.7).
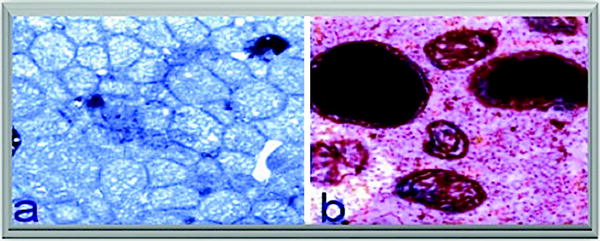
Fig. 12.7
Mitochondrial defects. (a) Increased mitochondrial number in cardiac muscle and (b) giant mitochondria with electron-dense deposits in skeletal muscle of a patient with mitochondrial cardiomyopathy
Enzyme immunostaining is a useful adjunct to evaluate the relative amounts of specific enzyme activity in muscle fibers and specific enzyme content, e.g., cytochrome c oxidase [76]. Moreover, immunohistochemical studies of skeletal muscle biopsies of children with COX deficiencies have been used to distinguish between mtDNA- and nuclear DNA-specific defects, based on the levels of mtDNA-encoded subunits COXI and COXII relative to changes in nuclear DNA-encoded subunits COXIV and COXVA [77].
Biochemical Analysis
Using either crude tissue homogenates or isolated mitochondria, the specific activity levels of each individual respiratory complex and/or bi-complexes (I + III, II + III) or respirasomes (see Chap. 18) can be evaluated. Mitochondria-based diseases such as MELAS and MERRF most often exhibit deficiencies in complex I and complex IV activities [78, 79]. Deficiencies in either complex V, complex IV, or in pyruvate dehydrogenase (PDH) activities have been described in Leigh syndrome [12, 80–82]. Complex IV deficiency has also been described in isolated cases of pediatric cardiomyopathy [83] and is the most common recognized respiratory chain defect in childhood. Infants with histiocytoid cardiomyopathy, which is characterized by cardiomegaly, ventricular tachycardia, and frequently sudden death occurring within the first 2 years of life, harbor defects in respiratory enzyme activities including complexes III [84] and IV [85]. Moreover, in children with DCM, reduced levels of specific cardiac respiratory chain enzyme activities were frequently found which ranged from 35 to 70% of age-matched control values [86]. Reduced activity levels of complex III were most often observed although cases exhibiting reduced activities of complexes I, IV, and V were also noted. Other studies using biopsied cardiac tissue from children with HCM found reduced complex I and IV activities [87]. Why specific mtDNA-encoded enzymes are affected, while others are unaffected, is not known.
MtDNA Analysis
As previously noted, a number of mtDNA mutations have been reported to play a role in the pathogenesis of cardiomyopathies. Defects in mtDNA identified thus far include specific point mutations in mitochondrial tRNA genes and in several of the 13 protein-encoding mitochondrial structural genes.
Other mtDNA abnormalities include increased abundance of specific large-scale mtDNA deletions in cardiomyopathic tissues although their pathogenic role remains to be established (Fig. 12.8). Additional types of mtDNA rearrangement (including tandem duplications of sequence) and defective genetic loci (including mitochondrial ribosomal RNA genes) have also been reported [88, 89]. Distinguishing pathogenic mutations from polymorphic variations in nucleotide sequence is not simple. Moreover, a number of mtDNA sequence changes previously characterized as polymorphic variants have been found to be associated with an increased incidence of cardiomyopathy; these include both homoplasmic and heteroplasmic changes in the conserved mitochondrial D-loop regulatory region (e.g., nt 16093, 16168, 16186, and 16189) [53, 90, 91].
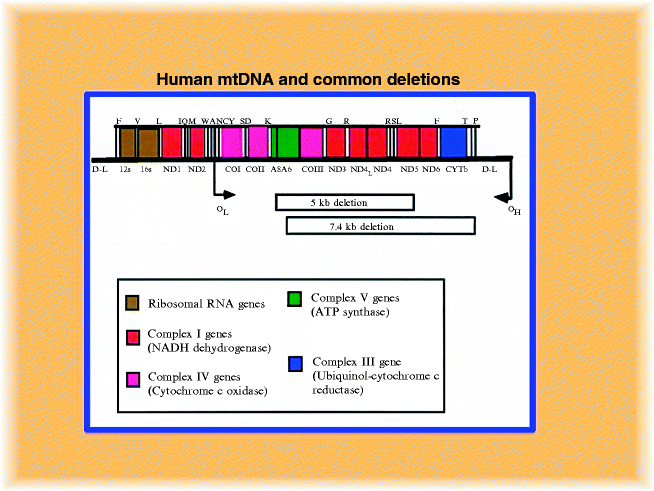
Fig. 12.8
Linear representation of mitochondrial DNA and common deletions. It shows the location of all 13 protein-encoding genes (ND1-ND6, COI-COIII, cytb and ATP6, and ATP8), 22 tRNAs identified by their cognate amino acid using single-letter code(F, V, L, I, Q, M, W, A, N, C, Y, S, D, K, G, R, H, S, L, E, T, P), the two rRNA genes (12S and 16S), and the noncoding D-loop region (D-L). Position of pathogenic mtDNA mutations in tRNA genes including leu, lys, ile, and gly is shown
Mitochondrial tRNA Mutations
Defects in mitochondrial tRNAs, which play a key role in protein synthesis, are involved in the genesis of multisystemic diseases with associated cardiomyopathy, including MELAS [92], MERRF [93], and LS [94].
Parenthetically, a double-point mutation in mtDNA has been found in the proband, a 23-year-old woman with MERRF harboring 8356 T > C and 3243A > G transitions in mitochondrial tRNA genes and three relatives patients whose phenotypes were MERRF, MERRF/MELAS overlap syndrome, and asymptomatic carrier. It was suggested that the course of the phenotype of this family begins with MERRF and followed by MELAS. This double mutation was heteroplasmic in blood in each case but with different rates, the mt.8356 T > C appeared homoplasmic and the mt.3243A > G heteroplasmic in muscle of the two cases studied. No other mutations were detected in the total mtDNA sequence in this family [95]. To the best of our knowledge, no cardiac pathology has been so far identified.
In our and other institutions, children with cardiomyopathy are screened for evidence of tRNA mutation using a variety of techniques such as single-strand conformation polymorphism (SSCP) analysis [96], restriction enzyme digestion [97], and automated DNA sequencing [98]. Point mutations in tRNA genes (e.g., tRNALeu, tRNAIle, tRNAVal, tRNALys, tRNAGly, and tRNAcys) have been associated with CM and HF [64, 66, 68, 99–103] (a number of which are shown in Table 12.3). Molecular data concerning the location of these mutations, biochemical findings concerning the enzymes affected, the inheritance pattern, the relative proportion of mutant relative to wild-type alleles (heteroplasmy), and pertinent clinical findings are also presented in Table 12.3. For certain mutations, e.g., 3243 in tRNALeu and 8344 in tRNALys, clinical manifestations are often delayed occurring primarily in adolescents or adults. On the other hand, atypical, early presentation of cardiomyopathy has been reported in cases with mutation at nt 3243 [104] and in a case of fatal infantile histiocytoid cardiomyopathy with a sporadic nt 8344 mutation in heart and liver [85]. For other less frequent tRNA mutations, particularly those associated with fatal infantile cardiomyopathy (e.g., 4269 tRNAIle, 4317 tRNAIle, 4320 tRNAIle, and 3303 tRNALeu), given the limited number of cases reported thus far, it is presently unknown whether or not the clinical outcome will invariably be the same. The majority of the aforementioned studies detected tRNA mutations using mtDNA derived from biopsied skeletal muscle; in some instances, the amount of the mutant allele is high enough to be detected in blood as well [105]. However, more often, analysis of the patient’s blood has provided limited information since hematopoietic cells may lose mtDNA mutations [1, 7]. Moreover, different quantities of both normal and mutant alleles may be found in the patient (i.e., heteroplasmy), an intracellular mixture of wild-type and mutant genomes which may be different dependent on the tissue examined. This is in contrast with situations where only one allele or the other is found (i.e., homoplasmy). Only when the percentage of mutant tRNA genes increases above a certain tissue-specific threshold does protein synthesis become inhibited, resulting in decreased activity levels of specific respiration enzymes and abnormal cellular phenotypes [1, 2]. Young individuals (<20 years of age) appear to have higher thresholds requiring as much as 95% of mutant mtDNAs in order to express the disease phenotype.
Table 12.3
tRNA mutations in mitochondrial cardiomyopathy
Gene | Site | Freq | HOM/HET | Signs and symptoms | References | Biochemical findings |
|---|---|---|---|---|---|---|
leu | 323 A>G | NUM | HET | MELAS, RRF, HCM | [46] | Defects in I + IV activities and protein translation |
leu | 320 A>G | LIM | HET | Dypsnea, CM, tachycardia | [52] | I + IV defects |
leu | 333 C>T | LIM | HOM/HET | FICM myopathy | [4] | Unknown |
ile | 430 A>G | LIM | HOM/HET | Dyspnea, RRF, HCM | [14] | No defects in muscle or heart |
ile | 4317 A>G | ND | ND | FICM | [5] | IV defect |
ile | 4320 C>T | LIM | HET | FICM seizures | [53] | No defects in OXPHOS |
ile | 4269 A>G | LIM | HOM/HET | Deafness, epilepsy, RRF at 18 years | [6] | IV defect |
lys | 8344 A>G | NUM | HET | Leigh, MERRF, HistCM | [47] | I and IV defects |
lys | 8296 A>G | LIM | HET | Fatal HCM | [66] | ND |
lys | 8363 G>A | LIM | HET | Hearing loss, ataxia, HCM | I, III, and IV defects; high II activity | |
gly | 9997 T>C | LIM | HET | Dysrhythmia, HCM | [47] | I, III + IV defects |
cys | 5783 G>A | LIM | HOM | Myopathy, HF at 8 years, deafness | [67] | I, II/III, and IV defects |
Moderately and severely deleterious mtDNA mutations are likely to be heteroplasmic, while mildly deleterious mtDNA mutations are homoplasmic [106]. For example, the 3303 tRNALeu mutation was described as homoplasmic in the proband and a close relative both who died of fatal infantile cardiomyopathy; however, maternal relatives with milder symptoms were heteroplasmic for this allele [4]. A homoplasmic mutation at 12192 in tRNAHis has been reported in several patients with DCM [107]. A pathogenic mutation in tRNAIle at nt 4300 was homoplasmic in all samples from several unrelated children affected with severe HCM as well as their maternal relatives [108]. Such findings suggest that caution is warranted in assessing pathogenicity as a function of heteroplasmy.
Detection of specific tRNA mutations has been made relatively simple by the use of restriction fragment digestion of PCR-amplified products containing the gene sequence of interest. Defective mtDNA genes can be characterized by either a loss or a gain of a restriction enzyme site. For the analysis of sequence changes which do not alter natural restriction sites, a restriction site can be created using “mismatched” primers which will distinguish between the presence of the wild-type and mutant alleles [109]. All of the point mutations in tRNA noted in Table 12.3 have been analyzed by restriction digestion. This approach has made possible the detection of each of the defective alleles described above using minimal tissue, limited technology, and resources. Similar diagnostic tests can also be performed with genes, e.g., beta-myosin heavy chain (β-MHC) and acyl-CoA dehydrogenase in which a number of specific mutations have been identified that are associated with a poor clinical outcome in patients with HCM [110]. These molecular genetic tests in combination with an analysis of pedigree/inheritance (e.g., an autosomal dominant pattern for β-myosin defects) can be used in excluding these defects, which are particularly common in cases of familial HCM [110, 111], in the differential diagnosis of MCM.
A number of mitochondrial tRNA mutations have been reported whose association with cardiomyopathy remains to be confirmed. For instance, a heteroplasmic mutation in the tRNAPro at 15990 converting the tRNAPro anticodon to that of tRNASer has been reported in a child with a “pure myopathy” characterized by progressive weakness of the extremities [112]. While mitochondrial respiratory complex activity levels in the patient’s biopsied skeletal muscle were severely decreased, there was no evidence of cardiac involvement.
Mutations have also been found in the anticodon stem and loop of tRNAThr at 15923 and 15924 in 2 unrelated cases of fatal infantile respiratory failure [113] and in tRNAThr at 15928 in several cases of cardiomyopathy [114]. Although the pathogenicity of these mutations has been called into question, since the 15924 tRNAThr and 15928 tRNAThr mutations have been observed in control tissues [115], the mutant allele at nt 15924 has been confirmed in patients with cardiomyopathy rather than in normal subjects [116].
Mitochondrial Structural Gene Mutations
Although rarely present, point mutations in moderately and highly conserved amino acid residues associated with MCM have been identified in approximately half of the 13 mtDNA-encoded proteins, including ATPase6, cytb, COXI, COXII, ND3, ND5, and ND1, and are discussed in more detail below.
ATP Synthase (ATPase6)
Leigh disease (LD) is a rapidly progressive neurodegenerative childhood disease with variable clinical manifestations including seizures, ataxia, basal ganglia lesions, and muscle weakness that can be associated with cardiomyopathy [12, 80]. A mutation in ATPase6 has been reported in several independent cases of Leigh syndrome with associated cardiomyopathy. The ATPase6 mutation is a T → G transversion at np 8993 converting a highly conserved hydrophobic leucine to arginine [117]. In biochemical studies with cultured lymphocytes derived from patients with LD, a pronounced defect was noted in the proton channel of the H+-translocating ATP synthase, thus inhibiting ATP generation from OXPHOS [118]. Abnormal activity of complex V was also found in the heart and skeletal muscles of an unrelated individual with Leigh cardiomyopathy containing the same mutation [80]. This mutation has been shown to present itself heteroplasmically in maternal relatives, while the patient had >95% mutant mtDNA in several tissues, e.g., heart, brain, and skeletal muscles. The relative concentration of the mutant allele correlates with the clinical severity of the disease.
A severe phenotype (“true” Leigh syndrome) occurs with a higher percentage of mutant genomes; lesser amounts of the mutant allele are associated with milder symptoms often termed NARP syndrome (neurogenic muscle weakness, ataxia, and retinitis pigmentosa) [119]. A second heteroplasmic, maternally inherited mutation located at the same position 8993 (T → C) resulting in a leucine → proline change has also been reported [107]. Although the T8993C mutation is generally considered to be clinically milder than the T8993G mutation when the level of heteroplasmy exceeds 90%, progressive neurodegeneration has been reported [120]. Mutations at a variety of other genetic loci can also cause Leigh syndrome, e.g., point mutations at several sites including the mitochondrial tRNALys gene [94], nuclear genes encoding subunits of pyruvate dehydrogenase [82], the flavoprotein subunit of complex II [121], and COX assembly factor SCO2 [52].
Cytochrome b
Several mutations in the cytochrome b (cytb) gene have been identified in children with cardiomyopathy. The first, an A → G mutation at nt 14927 in cytb, converting a highly conserved threonine to an alanine residue, was found in the heart mtDNA of a 1-year-old male with marked cardiomegaly and HF [11]. Also, a mutation at nt 15236 converting a highly conserved isoleucine to a valine at residue 164 has been reported in 2 unrelated patients [49]: a 7-year-old female with familial DCM and a 19-year-old male presenting with HCM. However, data concerning the extent of heteroplasmy, inheritance pattern, and respiratory enzyme activity analysis were not reported in these cases. Another cytb mutation, a C → A at nt 15452 in cytb, converted a leucine to an isoleucine at residue 236 [122]. This mutation was heteroplasmic, present in 7 of 33 cases of DCM screened and not found in controls (18 individuals), with four of the 7 mutant alleles detected in infants under 2 years of age. In these patients, there was a striking reduction in cardiac complex III activity with levels ranging from 0 to 40% of the control value. Since this mutation does not result in changing a highly conserved amino acid residue, its presence in several cases of Leber’s hereditary optic neuropathy (LHON) was previously considered nonpathogenic [123]. Nevertheless, to rigorously prove the pathogenicity of this mutation, its relationship to both the defective complex III activity and to the cellular phenotype will require the use of cybrids in which defective mitochondria containing the mutant gene are introduced into cultured cells by cell fusion [124].
In a child with COX deficiency and displaying severe histiocytoid cardiomyopathy, a mutant allele in cytb at nt 15498 was detected [125]. This mutation results in the replacement of a neutral amino acid (glycine) with an acidic one (aspartic acid) at a highly conserved residue (251), altering the interactions of cytochrome b with other subunits thereby affecting complex III assembly and proton transfer function [125, 126]. Another study identified a cytb mutation at nt 15243 in a patient with severe HCM and complex III deficiency. This mutation changes a highly conserved glycine to a glutamic acid residue situated within the cd2 helical region of cytb in close proximity to the hinge region of the Rieske iron–sulfur protein and was found to profoundly alter the subunit interactions, conformation of the bc1 complex, and complex III stability and activity [127, 128]. A cytb mutation at nt 15508 in a highly conserved aspartic with a glutamic acid residue was found in a female infant with DCM in association with marked complex III deficiency [114].
It is noteworthy that a relatively large number of pathogenic mutations in cytb have been found in cardiomyopathic tissues. While a subset of these mutations represent either neutral amino acid substitutions or likely polymorphisms, it remains to be seen whether there is some additive or synergistic effect of these mutations on mitochondrial function. These findings also imply that cytb represents a hitherto uncharacterized “hotspot” for mtDNA defects some of which could play an important role in the pathogenesis of cardiomyopathy adding it to a list that includes several mitochondrial tRNA genes (e.g., tRNALeu, tRNAIle, and tRNALys) and ATPase6 [1, 2, 7].
Mutations in COX and ND Subunits
Obayashi et al. [11] found a mutation at COII at nt 7673 converting a highly conserved isoleucine to valine at residue 30 in a 1-year-old patient with cardiomegaly. Ozawa et al. [49] reported a mutation in COI at nt 6521 causing a change from highly conserved isoleucine to methionine at residue 206 in a 7-year-old female with DCM. However, lack of information concerning allele heteroplasmy and enzymatic data precluded a complete evaluation of these mutations’ pathogenicity.
Several potentially pathogenic mutations have been reported in mtDNA genes encoding complex I subunits. One in ND5 at nt 13258 converted a highly conserved serine to a cysteine residue in a child with HCM [11] and a second mutation in ND1 at nt 3394 converting a highly conserved threonine to a histidine residue in a child with DCM [49]. Another mutation in ND1 at nt 3310 has been described in a case of HCM in conjunction with type 2 diabetes [129]. As with the COX mutations described above, no biochemical data were available. A severe reduction of complex I activity was found in association with a mutation in ND5 at nt 14069, resulting in a serine to leucine transition in a female of 14 years old with DCM [75] [114].
MtDNA Depletion
Mitochondrial DNA depletion appears to be a potential cause of mitochondrial cardiomyopathy, and depletion of wild-type mtDNA may be the primary defect in patients with mitochondrial myopathy and respiratory enzyme defects. The basis for the respiratory enzyme abnormalities found in some cases with MCM can be explained as a result of either pathogenic mtDNA mutation or mtDNA depletion. These alterations may be tissue specific and autosomally transmitted [130–133]. Several nuclear loci have been identified as likely responsible for mtDNA depletion, although some drugs like azidothymidine (AZT), which inhibits both the viral DNA polymerase and mitochondrial DNA POLG, abrogating mtDNA replication, are known to elicit mtDNA depletion and associated myopathies [57]. Also, autosomal-recessive mutations in enzymes that play a role in mitochondrial nucleotide metabolism (e.g., thymidine kinase, thymidine phosphorylase, and deoxyguanosine kinase) have been identified in a subset of patients with mtDNA depletion and represent an example of “dual genome” disorders. To confirm the pathogenic role that mtDNA depletion play in cardiomyopathy a systematic and accurate estimation of total undeleted mitochondrial genomes and their relationship to cardiac respiratory enzyme defects is needed. Dual-genome oligonucleotide array-based comparative genomic hybridization has been applied to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes by Chinault et al. [134]. This custom array can reliably detect mitochondrial DNA deletion with identification of the deletion break points and the percentage of heteroplasmy. Simultaneous detection of copy number changes in both nuclear and mitochondrial genomes potentially makes this dual genome of significant value in the diagnoses of mitochondrial DNA depletion syndromes.
Mitochondrial Changes in Congenital Heart Defects
Single-gene mutations have been implicated in the pathogenesis of CHD. These mutations are more common than previously thought and may be present in an number of genes involved in cardiac structure and function, including extracellular matrix proteins, metabolic enzymes and membrane transporters, fatty acid and mitochondrial biosynthesis, cardiac OXPHOS metabolism, sarcomeric structural and contractile proteins, as well as nuclear transcription factors, which control myocardial gene expression and developmental programming. In addition, abnormalities in the structure and function of mitochondria have been found in a significant number of children with CHD, and several subclasses of these mitochondrial defects will be discussed in this section.
Structural and Functional Cardiac Defects
Ultrastructural changes and abnormal mitochondrial number have been found in a variety of CHD, including ventricular septal defect, PDA, and right ventricular dysplasia [135, 136]. In addition, DiGeorge/velocardiofacial syndrome, which can present with tetralogy of Fallot, results from a large chromosomal deletion ranging from 1.5 to 3 Mb in chromosome 22 excising over 30 genes, some of which appear to be involved in mitochondrial function [137]. The loss of the gene for the mitochondrial citrate transporter was initially proposed as contributing to the distinct phenotype of this syndrome [137]. However, recent evidence implicated the deletion of the gene encoding the T-box transcription factor TBX1 in the cardiovascular abnormalities present in velocardiofacial/DiGeorge syndrome [138].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


