Mesenchymal Tumors of the Pleura
Adina Paulk, M.D.
Allen P. Burke, M.D.
Juliana Magalhães Cavalcante, M.D.
Fabio R. Tavora, M.D., Ph.D.
Solitary Fibrous Tumor
Terminology
Solitary fibrous tumor (SFT), previously called localized fibrous tumor,1 is a low-grade mesenchymal neoplasm. Tumors that were previously called hemangiopericytomas are now considered SFTs, except those in the meninges, which still retain the designation. The term “hemangiopericytoma-like” is still frequently used to describe the characteristic tumor vasculature of these lesions.
Clinical Findings
SFT occurs in adults, most frequently in the sixth decade, but has been reported in patients from their teens to their nineties.1 There is no gender predilection. SFT represents <5% of primary pleural neoplasms, with an estimated incidence of 2.8 cases per 100,000 individuals.2
Many SFTs are asymptomatic and discovered incidentally. Larger tumors cause symptoms of cough, dyspnea, and chest pain. Occasionally, patients become hypoglycemic related to tumor production of an insulin-like growth factor.3
Radiologic Findings
On imaging, SFTs are usually sharply demarcated pleural-based soft tissue masses with no chest wall abnormality. Occasionally, they are diagnosed within the parenchyma without pleural attachment.4 Positron emission tomography (PET) usually shows increased avidity in malignant versus benign tumors, although there is overlap.5
Gross Findings
SFTs are well-circumscribed, solid gray, homogenous masses, frequently pedunculated, and are often >10 cm (so-called giant SFTS). Cystic necrosis, hemorrhage, and calcification are occasionally present (Fig. 96.1).
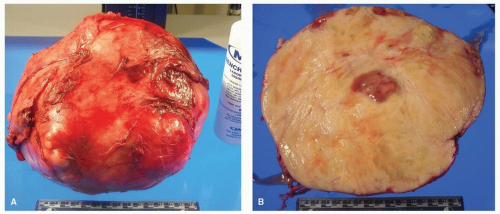 FIGURE 96.1 ▲ Solitary fibrous tumor, gross. A. The tumor is over 10 cm in size and partly covered by pleura. B. On cut section, there may be areas of cystic change with necrosis. |
Microscopic Findings and Differential Diagnosis
SFTs have a distinctive morphology, with uniform fibroblastic spindle cells, varying cellularity (cellular and paucicellar areas), variable fibrosis, and branching “hemangiopericytoma-like” or “staghorn” vessels (Figs. 96.2 and 96.3). The differential diagnosis includes synovial sarcoma, which is usually more cellular and mitotically active and lacks background fibrosis, and fibromatosis, which generally occurs in the soft tissue of the chest wall. Occasionally, growth into the lung parenchyma can result in nests and papillae of hyperplastic pneumocytes, mimicking sclerosing pneumocytoma (Fig. 96.4).
Immunohistochemical and Molecular Findings
The large majority of lesions are CD34 positive (Fig. 96.5), but this is nonspecific, and occasional SFTs do not express this marker. Positive staining for Bcl2 and CD99 is also nonspecific (expressed, e.g., in some synovial sarcomas) but can be helpful in evaluating small biopsies.6 Much more specific is STAT6, which is positive in >95% of cases.7
Criteria for Malignancy
The classic study of England et al. determined that a cutoff of >4 mitotic figures per 10 high-powered fields separated benign tumors (without any recurrence on follow-up) to aggressive neoplasms, 65% of which recurred.1 This criterion is still used, with a variation of 2 mm2 instead of 10 HPF.2 Using this criterion, ˜10% of SFTs are malignant.
Although recurrences are common in malignant SFT, distant metastases occur rarely.1,4,6 A single series showed a relatively high rate
of distant metastasis.8 The histologic appearance of malignant lesions include, in addition to increased mitotic activity, increased cellularity, hemorrhage, necrosis,1 and areas with the appearance of sarcoma.9
of distant metastasis.8 The histologic appearance of malignant lesions include, in addition to increased mitotic activity, increased cellularity, hemorrhage, necrosis,1 and areas with the appearance of sarcoma.9
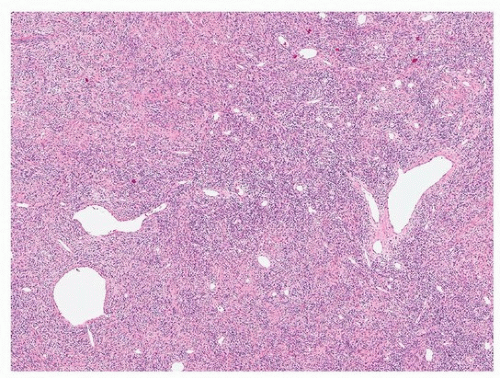 FIGURE 96.2 ▲ Solitary fibrous tumor. The “hemangiopericytomalike” vascular pattern is evident. |
Recurrences have shown decrease in CD34 expression, and malignant SFTs are more likely to be p53 and p16 positive than benign SFT.6,10,11 Ki 67 has been reported as increased as would be expected in high-grade tumors (Fig. 96.6).6,11 Ki 67 index did not correlate with recurrence in one study, which did show a correlation between recurrence and p16 expression.6
A three-tiered grading system has been proposed for SFT of the lung.4 Low-grade SFT has <4 mitoses per 10 HPF (corresponding to “benign” SFT), intermediate has grade 5 to 10 mitotic figures per 10 HPF with increased cellularity, and high-grade tumors show undifferentiated pleomorphic sarcoma (Fig. 96.6). Another study suggested a grading system based on bad prognostic factors (parietal pleura vs. visceral pleural, sessile vs. pedunculated, >10 cm vs. <10 cm, necrosis, and mitotic figures >4 per HPF).12
A single malignant SFT with heterologous elements (osteosarcoma) has been described.13
Fibromatosis
Terminology
Fibromatoses are relatively rare tumors derived from fascial or musculoaponeurotic structures. The term “desmoid tumor” is synonymous and favored by surgeons.
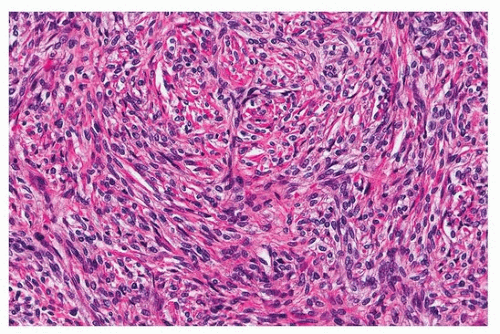 FIGURE 96.3 ▲ Solitary fibrous tumor. Highest magnification demonstrates collagen encircling individual tumor cells. |
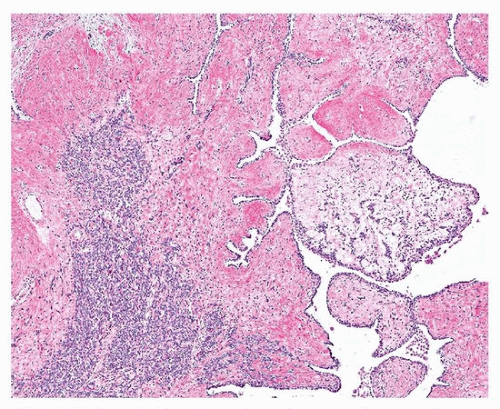 FIGURE 96.4 ▲ Solitary fibrous tumor. There can be areas of dense collagen, and, when invading the lung parenchyma, pneumocyte inclusions or papillae can be seen that are TTF-1 positive (not shown). |
Clinical Findings
Approximately one-third of fibromatoses occur on the trunk, usually the chest wall, breast, or axilla.14,15,16 Most patients are young or middleaged adults with visible subcutaneous swelling that frequently invades bone on imaging.17 A history of prior surgery or trauma is elicited in 10% to 20% of patients.15 Occasionally, chest wall or mediastinal fibromatoses can involve the pleura or lung, mimicking a primary lung or pleural tumor.18,19,20
Primary pleural fibromatoses are uncommon and usually arise on the parietal pleura. The largest series comprises four cases.21 Patients ranged from 16 to 66 years, and in two patients (50%), there was an antecedent history of trauma.21
Gross and Microscopic Findings
Pleural fibromatosis are typically large (mean 12.5 cm) and exhibit a bosselated, firm, white, cut surface.21 Fibromatoses of the chest wall can reach giant proportions.24
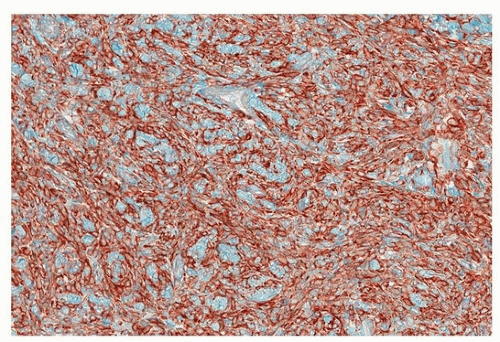 FIGURE 96.5 ▲ Solitary fibrous tumor. CD34 is usually diffusely positive but is not as sensitive or specific as is STAT6. |
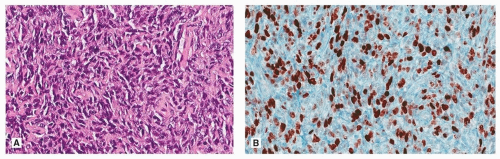 FIGURE 96.6 ▲ Malignant solitary fibrous tumor. A. There is increased cellularity and cellular pleomorphism. The appearance is similar to undifferentiated pleomorphic sarcoma. B. Immunohistochemical staining for Ki 67 shows a high proliferative rate. |
The histologic features of intrathoracic desmoid tumors are similar to those of desmoid tumors elsewhere in the body (Figs. 96.7 and 96.8). Pleural fibromatosis infiltrates the adjacent fat and skeletal muscle (Fig. 96.9). Tumor cells are immunoreactive for vimentin, desmin, smooth muscle actin, and muscle-specific actin, express β-catenin, and are negative for neural markers such as S100 protein and CD34.
A report has described mesothelial rests in axillary lymph nodes draining a chest wall fibromatosis.25
Differential Diagnosis
Desmoid tumor should be considered in the differential of localized fibrous tumor of the pleura and neurogenic tumors of the chest wall adjacent to the thoracic spine. The diagnosis is generally made easily by histologic examination. Immunohistochemical stains are helpful to exclude neural tumors (positive for S100 protein, negative for β-catenin) and SFT (positive for CD34, stat6, and negative for β-catenin).
Prognosis and Treatment
Complete resection with wide margins is the standard of care for fibromatoses, regardless of site. Recurrences occur in one-third of tumors in all sites. Because fewer than one-half of patients with positive margins experience recurrence, overly aggressive surgery is to be avoided.
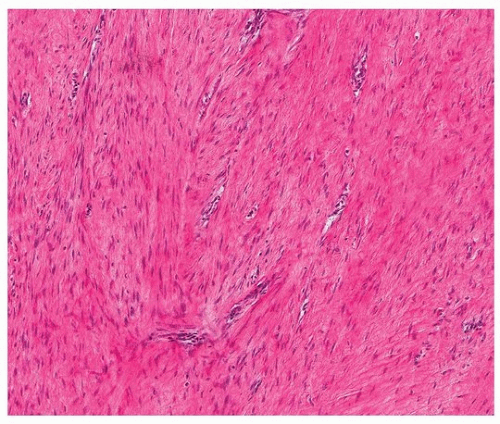 FIGURE 96.7 ▲ Pleural fibromatosis. An intermediate magnification shows dense fascicles of collagen with a low density of parallel-oriented fibroblasts. There are fairly regularly spaced thin-walled vessels. |
Factors associated with recurrence include tumor size larger than 5 cm, extra-abdominal tumor location, and incomplete resection.15 Residual disease can remain stable for up to 1 year.21
Treatment of nonresectable tumors includes radiation therapy, steroid injection,26 antiestrogens, aromatase inhibitors,27 antiprogesterone drugs,28 and tranilast.29
The majority of chest wall fibromatoses can be completely excised, with a low rate of recurrence (17%).17
Pleuropulmonary Synovial Sarcoma
Introduction
Synovial sarcoma (SS) is a malignant soft tissue tumor of uncertain differentiation and may be monophasic or biphasic, with epithelial and mesenchymal components. SS of the lung typically involves the pleura with extension into the parenchyma (pleuropulmonary synovial sarcoma), with 75% of tumors showing a pleural base by computed tomography scanning.30 Pleuropulmonary synovial sarcoma is uncommon; there were fewer than 100 cases reported up until 2007.30
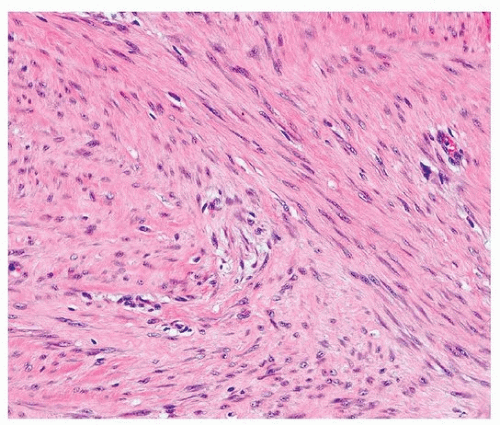 FIGURE 96.8 ▲ Pleural fibromatosis. A higher magnification shows bland fibroblasts and collagen. |
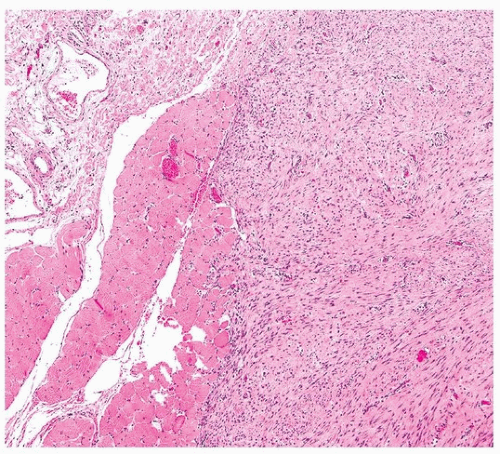 FIGURE 96.9 ▲ Pleural fibromatosis. The tumor is infiltrating skeletal muscle, at the left. |
Clinical Features
The mean age range at presentation is 42 to 47 years (overall age range, 9 to 77 years).30,31 There is no gender predilection.30,31,32,33 Typical signs and symptoms include chest pain (24% to 80%), dyspnea (8% to 36%), cough (8% to 33%), and hemoptysis (20% to 25%).34 Pleural SS can be aggressive with almost half of patients dead of disease (with a mean of 18 months).34
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


