Chapter 12 Medical Management of Vascular Disease Including Pharmacology of Drugs Used in Vascular Disease Management
Arterial disease is the leading cause of death and significant morbidity in the United States and throughout the world. The American Heart Association estimates that 80 million (36.3%) Americans have cardiovascular disease, leading to 864,500 deaths annually.1a Patients with peripheral arterial disease (PAD) compose a significant proportion of this group, including 795,000 Americans who will have strokes each year. Stroke itself is the third leading cause of death in the United States, with an estimated 143,600 patients dying each year. Those who survive often have significant neurologic deficits that can become major social and economic burdens to the patients and their families.
PAD is a significant public health issue because of the need for extensive long-term care for patients with these serious disabilities and because much of atherosclerosis and therefore PAD is preventable or diminishable by avoiding tobacco, fatty foods, and taking medications regularly to control hypertension, diabetes, and hyperlipidemia. Although we now have excellent diagnostic modalities that can help to identify patients at risk for atherosclerosis, along with well-planned preventative strategies, patients are often still reluctant to make the necessary lifestyle modifications. It is estimated that for 2009, the total direct and indirect cost of cardiovascular diseases and stroke in the United States was $475.3 billion.1a
Atherosclerosis Basic Principles and Medical Management
Risk Factors for Atherosclerotic Disease and Modification Strategies
Many theories and causes of atherogenesis have been postulated and studied, and they have led to a handful of clear risk factors for atherosclerosis. These risk factors have long been recognized as the framework for a preventative strategy for cardiovascular diseases. The identification of specific cardiovascular risk factors that led to the concept of risk factor modification arose from the findings of the Framingham Heart Study of the 1960s.14 A risk factor can be defined simply as an entity that can be identified early in the disease course of an individual or group of individuals and confers an increased risk of disease development. An essential part of disease prevention in the context of identification of risk factors is the ability for modification of that specific risk factor, usually in the form of behavioral or pharmacologic manipulation.
Cardiovascular disease is increasingly recognized as the largest growing burden of disease for health care systems.15 Though progress in treatment regimens and surgical outcomes once atherosclerotic disease is established has improved morbidity and mortality from cardiovascular complications, there has been a shift in emphasis toward the development of effective clinical guidelines for prevention and modification at an earlier stage in the disease process. Box 12-1 lists the most common risk factors for atherosclerotic cardiovascular disease.
Smoking
Smoking is the greatest contributor to atherosclerotic cardiovascular disease and is the number one cause of preventable deaths in the United States annually.16 There has been an increase in the number of smokers despite extensive antismoking campaigns, and smoke-related deaths continue to rise particularly in the developing world.
A dose-related phenomenon has been described for cigarette-smoking that correlates with increased rates of coronary events, ischemic strokes, and peripheral vascular disorders.9 Despite this dose effect, complete smoking cessation has been demonstrated to be the only significantly effective approach to reducing health risks associated with smoking. Smoking cessation remains a pivotal part of cardiovascular disease prevention. However, initiatives aimed at improving public awareness of the deleterious effects of ischemic heart disease, peripheral vascular disease including amputation, and cerebrovascular events have met with limited success. As much as one third of cardiovascular mortality can be prevented by abstinence from smoking, an effect that has not yet been realized by pharmaceutical risk-factor management.17
Attitudinal approaches to smoking cessation have been well described in the addiction literature and can be classified into five main stages: precontemplation, contemplation, preparation, action, and maintenance. These five stages are often referred to within the construct of the transtheoretical model of health behavior change.18
Diabetes Mellitus
Coronary artery disease is the principal cause of death in diabetic patients and rivals smoking in contribution to cardiovascular mortality.19 The rate of coronary and peripheral arterial disease approximately doubles in patients with a diagnosis of diabetes.20 The length of time and severity of diabetic control are strong predictors of atherosclerotic events and have been correlated with the degree of peripheral arterial disease experienced by patients. The microvascular complications of diabetes are beyond the scope of this chapter but diabetic nephropathy, heralded by microalbuminuria, exacerbates large vessel changes imposed by insulin resistance and hyperglycemia.21 Atherosclerosis can be shown experimentally to be induced by insulin resistance preceding the development of a clinical diagnosis of diabetes and has been diagnosed in adolescents and teenagers as part of the metabolic syndrome.
Significant improvements in the glycemic profile and reduction in diabetic complications with prolongation of life expectancy can be achieved through behavioral modification of diabetes. Level 1 data from large, randomized national trials of monitored lifestyle modification demonstrate up to 30% reduction in frank diabetes with associated reduction in cardiovascular events. The addition of effective glycemic agents such as metformin, sulfonylureas, and thiazolidinediones further contributes to cardiovascular risk reduction. Current glucose targets for diabetic patients are listed in Box 12-2. A causal relationship has been described for long-term blood glucose control as assessed by the hemoglobin A1c (HbA1c) in the national United Kingdom Prospective Diabetes Study (UKPDS) with an increase in the risk of adverse cardiovascular events for each percentage point above an HbA1c level of 6.2%. The UKPDS recommendations for metabolic control of diabetic patients also focus on other parameters that are known to interact deleteriously with diabetes to increase cardiovascular risk such as hypertension and hyperlipidemia.22 Physicians now recognize the need for aggressive management of patients with the constellation of diabetes, hypertriglyceridemia, hypertension, and obesity, some of which will fit the definition of the metabolic syndrome.
Box 12-2
Current Target Guidelines for Diabetic Patients
• Fasting blood glucose <110 mg/dL
• Blood pressure <130/80 mm Hg (combination HCTZ/ACE inhibitor as first-line therapy)
• LDL < 100 mg/dL (<70 mg/dL considered if established coronary artery disease)
HCTZ, Hydrochlorothiazide; ACE, angiotensin-converting enzyme; LDL, low-density lipoprotein.
Hypertension
The prevalence of hypertension in the United States is estimated at one in three individuals and rising steadily. Part of the difficulty in managing hypertension is the racial disparity in prevalence, response to antihypertensive medications, and associated exacerbating factors such as renal disease and diabetes. High-risk groups include African Americans, those older than 60 years, and women.23 A dose phenomenon has been described for hypertension. In general, an elevation in blood pressure of 20 mm Hg systolic from a theorized normal of 120 mm Hg systolic confers a cardiovascular risk double that of the normotensive population. A working definition of hypertension is a systolic blood pressure greater than 140 mm Hg or a diastolic pressure greater than 90 mm Hg. Prehypertension can be defined further as blood pressure ranging between 120 and 139 mm Hg systolic and 80 to 89 mm Hg diastolic.24 Traditional views of hypertension primarily focused on diastolic dysfunction and elevation as more significant than the systolic component; however, this perspective has been shifted with more recent data indicating a greater risk for cardiovascular events and mortality in the face of systolic hypertension. Pulse pressure increases for a given systolic blood pressure have also received more attention as markers of impairment of vascular receptive relaxation and predictors of coronary risk over normal controls. Although hypertension is an independent risk factor for the development of coronary disease, it should be noted that it is much more potent when considered alongside other commonly associated risk factors, such as triglyceride profile, diabetes, and obesity. Almost two thirds of patients with hypertension have at least one other risk factor for cardiovascular disease; therefore treatment of hypertension should be ideally managed using therapies that are multivariate in effect, such as dietary modification or pharmacologic agents with benefits for both triglyceride and blood pressure profile.
The 2003 Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC-7) panel published guidelines that summarize management goals for the treatment of hypertension-associated risk factors.24 For example, hyperlipidemia may be more difficult to control when a combination of diuretic and β-blocking agents is used because of slight alterations in lipid metabolism as a side-effect of these medications. In the presence of diabetes, calcium-channel blockers, angiotensin-converting enzyme (ACE) inhibitors, and β-blockers are recommended. Lifestyle modifications are clearly identified with the following JNC-7 guidelines: weight reduction or maintenance of body mass index between 18.5 and 24.9 kg/m2; the Dietary Approaches to Stop Hypertension eating plan consisting of a diet rich in fruits, vegetables, low-fat dairy products, low sodium (ideally less than 100 mmol or 6 g of sodium chloride per day), reduced fat content, moderate alcohol consumption with less than 2 drinks per day for men and 1 drink per day for women and lighter weight individuals; and regular aerobic activity for at least 30 minutes most days of the week.
Dyslipidemia
Low-density lipoprotein (LDL) is firmly established in cardiovascular risk profiling as the major contributor to atherosclerotic disease and is found in abundance in atherosclerotic plaque. Higher serum levels of LDL correlate to a higher risk of cardiovascular disease—data borne of clinical studies that indicate LDL as the main risk factor for coronary disease.25 The Post-Coronary Artery Bypass Graft trial examined this relationship further and attempted to define a threshold target level for LDL.26 From this work, guidelines were established supporting intensive LDL treatment to a level less than 100 mg/dL to influence a favorable change in atherosclerotic plaque morphology (see Box 12-2).
Although LDL has been identified as a target for cardiovascular risk reduction, other lipid and lipoprotein abnormalities have been recognized in contributing to the overall risk of atherosclerotic disease. Elevated levels of very-low-density lipoprotein (VLDL), apolipoprotein B, and decreased high-density lipoprotein (HDL) are adverse markers for cardiovascular risk.27 Clinical evidence for the involvement of these lipoproteins is found in the acceleration of atherosclerotic build-up seen in patients suffering from inherited forms of dyslipidemia, such as familial hypercholesterolemia.
First-line therapies for reducing cholesterol, LDL, and VLDL and increasing HDL involve behavioral modification in the form of dietary changes.28 Animal fat, including meats and egg yolks, is a significant source of cholesterol, and goal strategies focus on reduction of cholesterol-rich products by at least 50%. Simply reducing body weight toward a goal body mass index will produce significant reductions in LDL levels and reduce the overall cardiovascular risk profile.
The main pharmacologic modality used to impair cholesterol metabolism is the hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (or statin medications). The mechanism of statin action occurs at the hepatocellular level to inhibit cholesterol synthesis in the liver. Statins are powerfully effective in reducing total body LDL levels (from 30% to 60% depending on dosage). Not surprisingly, these medications have become first-line drug therapy for patients with elevated lipid profiles in the absence of drug contraindications. Myopathy, heralded by a rise in creatine kinase, and transient elevation of hepatic aminotransferases are the most commonly quoted side effects of statin use. These derangements usually resolve with discontinuation of the medication.28 Statins along with four other medications used in the treatment of dyslipidemias (ezetimibe, niacin, fibric acid derivatives, and bile acid sequestrants) will be discussed.
Metabolic Syndrome
Over the past half century, a constellation of metabolic derangements has been seen more frequently occurring in association. Hypertension, diabetes mellitus, obesity, and dyslipidemia are the four entities most commonly described as part of the metabolic syndrome or colloquially known as syndrome X.29 According to the definition established by the 2001 Adult Treatment Panel-III of the National Cholesterol Education Program metabolic syndrome is diagnosed when three of the following criteria are present27:
1. Central obesity; waist circumference >102 cm (males) or >88 cm (females)
2. Fasting plasma glucose >6.1 mmol/L
3. Hypertension ≥135/85 mm Hg or the presence of antihypertensive medications
4. Dyslipidemia including triglycerides ≥1.7 mmol/L, HDL cholesterol <1.0 mmol/L (males) or <1.3 mmol/L (females).
The metabolic syndrome for many of the reasons highlighted earlier in this chapter is strongly associated with the development of cardiovascular disease portending an approximately 2.5 times risk of fatal cardiovascular events in the population. The single most effective treatment for the metabolic syndrome is loss of body weight either by nonsurgical or surgical means, which in almost all cases of dramatic weight loss leads to amelioration of all individual components.30
Emerging Novel Risk Factors
Elevated plasma homocysteine levels have been cited as a defined risk factor for the development of atherosclerotic coronary arterial disease in epidemiologic and clinical research studies.31 On a molecular basis, high levels of homocysteine have been demonstrated to occur with disruption of normal methionine metabolism. Homocysteine and related metabolites can be detected in abnormally high levels in the blood and have been linked to an increased risk of stroke, owing to carotid plaque buildup, as well as cardiovascular disease.32 Endothelial damage in association with an alteration of the normal coagulation balance has led to the hypothesis that elevated homocysteine levels directly influence atherogenesis in large vessels. Therapeutic options for individuals diagnosed with homocystinemia have centered on replacement of vitamin B12 and folic acid as a primary treatment with additional restriction of dietary intake of methionine in patients insensitive to vitamin B12. However, studies have not shown that lowering homocysteine levels decreases the risk of cardiovascular disease in these patients.33
Hs-CRP has recently been the subject of much attention with the results of the Justification of the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial, which was a large multinational, double-blind, placebo-controlled trial of more than 17,000 people.34 The trial was designed to observe the effect that treatment with a statin (rosuvastatin) had on individuals with normal lipid profiles but elevated hs-CRP levels. The study arose from the observation that statins have an antiinflammatory property and decrease hs-CRP levels in an effect that is independent from their cholesterol-lowering ability. The JUPITER trial demonstrated a reduction in both LDL and hs-CRP levels to around half of pretreatment and was terminated prematurely on the basis of this beneficial result. However, the benefit of a normal hs-CRP has not yet been firmly established as a treatment goal in cardiovascular risk profiling and is additionally confounded by the possible benefit of statin therapy in the face of normal lipid profile in certain populations. Nevertheless, hs-CRP remains an active and controversial area of cardiovascular risk-modification research, and its clinical role is yet to be formally determined.35
Surveillance and Secondary Prevention
For patients who progress to severe or acute cardiovascular disease, secondary prevention guidelines are well documented and rigorously studied. Many cardiovascular centers have established protocols for treating patients with established disease. One such example is the University of California–Los Angeles Cardiac Hospitalization Atherosclerosis Management Program (CHAMP) which focuses on using secondary prevention measures while patients are in the hospital in order to improve clinical outcomes.36 This program arose through the observation that although evidence-based guidelines for secondary risk prevention are widely disseminated, they are consistently underutilized. The CHAMP guidelines are summarized as follows:
1. Aspirin should be initiated (81 to 162 mg daily). In the presence of contraindications to aspirin, other platelet agents should be considered, such as clopidogrel. Combination therapy can be recommended in the setting of acute coronary syndromes or postrevascularization therapy.
2. Statin therapy should be initiated in all patients in the absence of contraindications and in all diabetic patients regardless of their lipid profile. Target levels of LDL should be <70 mg/dL, HDL >40 mg/dL, and triglycerides <150 mg/dL.
3. ACE inhibitors or angiotensin-receptor blockers should be commenced in the absence of contraindication regardless of the blood pressure or cardiac ejection fraction.
4. Beta blockade should be prescribed for all patients in the absence of contraindication.
5. Fish oil or omega-3 fatty acids should be commenced with dietary instruction for all patients.
6. Aerobic exercise programs that involve 30 to 60 minutes of moderately intense exercise at least five times per week should be prescribed.
7. Smoking cessation should be pursued including access to formal smoking cessation programs.
8. Before hospital discharge and at 6-week follow-up, cardiovascular lipid profile and liver enzymes should be checked and routinely thereafter at future follow-up appointments.
Pharmacology of Drugs Used in the Management of Vascular Disease
Anticoagulants (Table 12-1)
Heparin
Structure and Mechanism of Action.
Heparin is an anticoagulant composed of a heterogeneous group of straight-chain glycosaminoglycans with molecular weights ranging from 5 to 30 kD (mean, 15 kD).1 It is strongly acidic secondary to its high content of sulfate and carboxyl groups. Heparin is a naturally occurring substance excreted by mast cells and basophils in the process of clot formation. Standard, unfractionated heparin is derived commercially from porcine gut mucosa or bovine lung tissue. Heparin acts at multiple points within the coagulation system. Its major anticoagulant effect is via interaction with antithrombin III, leading to the inactivation of factor Xa and subsequent inhibition of the conversion of prothrombin to thrombin.2,3 Heparin further inhibits coagulation by inactivating thrombin, preventing the conversion of fibrinogen to fibrin.4 Heparin also prevents stable fibrin clot formation through the inhibition of fibrin stabilization factor. Heparin has no fibrinolytic activity and therefore does not lyse existing clots.
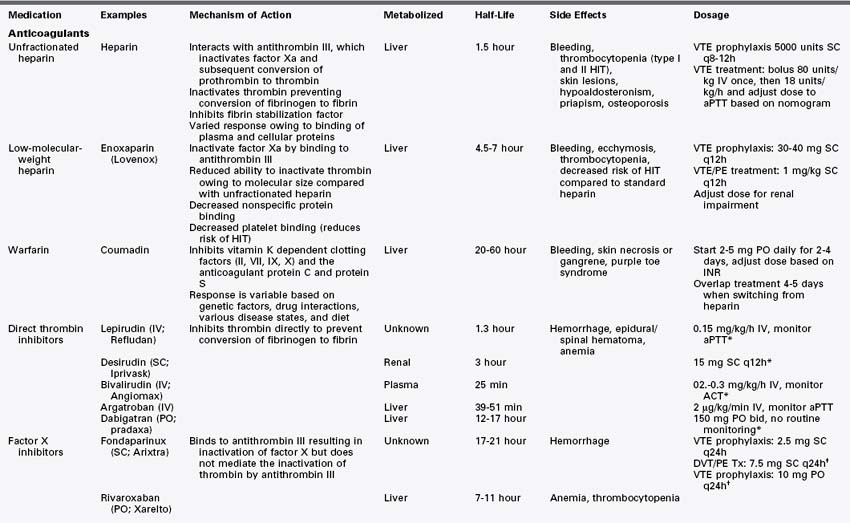

Heparin’s onset of action is immediate with intravenous injection. It can also be administered subcutaneously. Response to heparin is monitored by measuring the activated partial thromboplastin time (aPTT) and activated clotting time (ACT). An aPTT 1.5- to 2.5-fold greater than normal has been shown to prevent recurrent thromboembolism.5 Weight-adjusted nomograms have been shown to be useful in dosing.6
The anticoagulant activity of heparin varies greatly among patients. The heterogeneous clinical response is primarily due to nonspecific binding of heparin to variable concentrations of plasma and cellular proteins, limiting heparin’s bioavailability. This leads to marked variability of the anticoagulant response.2 There also appears to be natural inhibitors of heparin that can be released by sites of active thrombus.7 Furthermore, the biophysical limitations of the large heparin–antithrombin III complex can block receptors on thrombin to heparin cofactor 2, limiting heparin’s effectiveness.7
Clinical Use.
Heparin is indicated for intraoperative anticoagulation in vascular and cardiac surgery, for the prophylaxis and treatment of deep venous thrombosis, for the prevention of pulmonary embolism in surgical patients, and in patients with atrial fibrillation and embolization.2
Adverse Reactions.
Heparin therapy is associated with increased risk of bleeding. It also can cause skin lesions, including papules, plaques, and necrosis.8 Heparin therapy can lead to hypoaldosteronism,9 priapism,10 and osteoporosis.11 Thrombocytopenia is a known complication of heparin administration. Heparin-induced thrombocytopenia (HIT) can lead to thromboembolic complications, including skin necrosis, extremity gangrene, myocardial infarction, pulmonary embolism, and stroke.2
Heparin-Induced Thrombocytopenia.
Two major forms of HIT are recognized. Type I HIT is an early-onset, benign, reversible thrombocytopenia with no associated platelet antibodies. It is not immune mediated and is usually self limited without complications.12 Type II HIT is a more serious immune-modulated thrombocytopenia.13 It is caused by platelet immunoglobulin G antibodies that target platelet factor 4, a heparin binding protein, leading to platelet activation.14,15 Activation of platelets and endothelium and neutralization of heparin all contribute to a highly thrombogenic state.2 Patients with type II HIT have a high risk of developing thrombotic complications.2 Fortunately, most patients who develop HIT antibodies do not develop thrombocytopenia or thrombosis. Furthermore, almost all patients who develop mild to moderate thrombocytopenia during the first 4 days of heparin treatment do not have antibodies. Type II HIT should be considered a clinicopathologic syndrome with a combination of thrombocytopenia and associated clinical events, including thrombosis and confirmation of platelet antibodies.2 The management of HIT is aimed at preventing thromboembolic complications. All heparin should be discontinued and an alternative anticoagulant initiated.
Protamine
Structure and Mechanism of Action.
Protamine is composed of a heterogeneous group of low-molecular-weight proteins. These proteins are rich in arginine and are strongly basic. They occur naturally in the sperm of salmon and certain other fish species. The mechanism of action for heparin reversal is through electrostatic bonding. Heparin is highly acidic and forms a strong bond with the highly basic protamine molecules, forming an inactive complex.16
Adverse Reactions.
Too rapid administration can have serious side effects, including hypotension and anaphylaxis.17 Decreased blood pressure, pulmonary hypertension, shortness of breath, flushing, and urticaria have all been associated with rapid administration.18 Protamine should be administered slowly over 10 minutes, with a goal of 1 mg of protamine to neutralize every 90 units of heparin. Further dosing should be guided by coagulation studies.16 Severe allergies have been well documented causing transient systemic hypotension, anaphylaxix and severe pulmonary vasoconstriction. Patients with risk factors for a protamine reaction include those with fish allergies, vasectomy, and prior protamine exposure, including protamine insulin-dependent diabetics.19,20
Low-Molecular-Weight Heparins
Structure and Mechanism of Action.
Low-molecular-weight heparins (LMWHs) are collections of heparin molecules that have significantly lower molecular weights than standard, unfractionated heparin.21 LMWHs are derived from unfractionated heparin via chemical or enzymatic depolymerization. This produces fragments one third the size of heparin, with mean molecular weights of 4 to 5 kD (range, 1 to 10 kD). Similar to unfractionated heparin, LMWHs are heterogeneous in terms of both molecular size and anticoagulant activity.22 The LMWHs have distinct differences from standard heparin. LMWHs have reduced ability to catalyze the inactivation of thrombin, because the smaller fragments cannot bind to thrombin. However, LMWHs retain the ability to inactivate factor Xa. There is a reduction in nonspecific protein binding and subsequent improved predictability in dose-response relationships.22 LMWHs have an increased half-life compared with standard heparin. This is thought to be secondary to reduced macrophage binding. Similarly, there is reduced binding to platelets and a decrease in the incidence of HIT.22 LMWHs also have been associated with a reduction in bone loss compared with standard heparin.
Clinical Use.
The LMWHs have been examined extensively in the prevention of deep venous thrombosis in patients undergoing major abdominal surgery or knee and hip replacement surgery and in patients with restricted mobility.21 LMWHs have also been investigated for the treatment of acute deep venous thrombosis and pulmonary embolism.21 LMWHs are used in patients with acute coronary syndromes and unstable angina.23 The advantages of LMWHs over standard heparin are longer plasma half-life and a more predictable anticoagulant response, allowing for simple dosing and decreasing the need for laboratory monitoring.22 LMWHs are also commonly used to bridge patients to oral warfarin therapy.
Adverse Reactions.
Adverse reactions to LMWHs are similar to those associated with standard heparin. Bleeding, ecchymosis, and thrombocytopenia can all occur with LMWH administration. The incidence of HIT is decreased compared with standard heparin, but LMWHs can cause HIT, and patients should be monitored for this complication.24
Warfarin
Structure and Mechanism of Action.
Warfarin is a coumarin derivative that produces an anticoagulant effect through the inhibition of vitamin K–dependent coagulation factors (II, VII, IX, X) and the anticoagulant proteins C and S. Warfarin interferes with the conversion of vitamin K to 2,3-epoxide.25 Vitamin K is an essential cofactor for postribosomal synthesis of clotting factors, acting through the carboxylation of glutamine residues in the protein.26 Carboxylation promotes binding of vitamin K–dependent coagulation factors to phospholipid surfaces.25 Coumarins specifically block vitamin K epoxide reductase, preventing the carboxylation of the factors rendering them inactive.27 The vitamin K antagonists also inhibit carboxylation of the regulatory anticoagulant proteins C and S.27
Warfarin is a racemic mixture of two optically active isomers.25 Absorption from the gastrointestinal tract is rapid, reaching maximal plasma levels within 90 minutes. Warfarin has a half-life of 36 to 42 hours. Response is variable owing to certain genetic factors, drug interactions, various disease states, and diet. The anticoagulant effect can be overcome by low doses of vitamin K1, because vitamin K1 bypasses vitamin K epoxide reductase.25 Warfarin anticoagulation is monitored via measurement of the prothrombin time (PT). The PT reflects the depression of the vitamin K–dependent factors. PT measurement is laboratory dependent, and physicians should be aware of the specific method used to measure it at their institutions.
Clinical Use.
Warfarin is indicated for the prophylaxis or treatment of venous thrombosis and thromboembolism. It is also indicated for the prophylaxis or treatment of the thromboembolic complications associated with atrial fibrillation and cardiac valve replacement. Warfarin has been shown to reduce the risk of death, recurrent myocardial infarction, and thromboembolic events such as stroke or systemic embolization after myocardial infarction.25
Warfarin has been studied in terms of its usefulness in promoting the patency of infrainguinal bypass grafts.28–31 Although some studies suggest improved patency of infrainguinal bypass grafts with warfarin or a combination of warfarin and aspirin, the American College of Chest Physicians 8th Edition Guidelines for Antithrombotic Therapy for Peripheral Artery Occlusive Disease recommends against long-term anticoagulation in general for extremity reconstructions. The guidelines recommend vitamin K antagonists (VKAs) to not be routinely used for infrainguinal vein bypass, except in those patients at high risk of bypass occlusion and limb loss where VKAs plus aspirin is recommended. For infrainguinal prosthetic bypass, VKAs are not recommended to be used routinely.32 These conclusions are based on small improvements in patency in the face of relatively high rates of bleeding complications.
Adverse Reactions.
Warfarin therapy is associated with an increased risk of hemorrhagic complications. It also can cause necrosis and gangrene of skin or other tissues. It should be used with caution in patients with HIT, because it can lead to increased thrombotic complications early in the treatment of HIT.33
Direct Thrombin Inhibitors
Thrombin is the central enzyme in hemostasis. It is a serine protease that catalyzes the conversion of fibrinogen to fibrin. In addition, thrombin serves many other roles, including the activation of various coagulation factors, platelets, smooth muscle cells, fibroblasts, and endothelium.34 Thrombin is chemotactic, stimulates secretion of vasoactive proteins from platelets and inflammatory cells, and plays a role in angiogenesis and restenosis.34
Antithrombin III, the major regulator of thrombin, forms an irreversible complex with thrombin to block its active site.22 For years, this has been the main target for thrombin inhibition and anticoagulation via heparin. However, the use of heparin is limited by several major factors: (1) the development of HIT; (2) heparin’s inability to penetrate clot, leading to the release of active thrombin from clots; and (3) the variable anticoagulant response, owing to heparin’s propensity to bind to plasma proteins.35 The rationale for the development of direct thrombin inhibitors was to create a small molecule with site-specific thrombin inhibition.35 With an increased understanding of the detailed molecular structure of thrombin, site-specific agents have been developed with a high specificity for thrombin.36
Clinical Use.
Direct thrombin inhibitors have been studied in the treatment of HIT with thrombosis, the treatment of acute coronary events, and the prevention and treatment of deep venous thrombosis, pulmonary embolism, and stroke.34,36
Hirudin.
Hirudin, the first direct thrombin inhibitor, was originally isolated from the salivary gland of the medicinal leech Hirudo medicinalis after it was noted that leech saliva had anticoagulant properties.36 It is now produced via recombinant technology as lepirudin and desirudin. It is a 65– to 66–amino acid polypeptide. The amino terminus forms a tight bond with thrombin’s active site, and the carboxy terminus binds to the thrombin exosite-1 (fibrinogen binding site).37 Peak plasma levels are reached after parenteral administration in 20 to 30 minutes. Hirudin is rapidly cleared by the kidneys, having a half-life of 1 to 3 hours. Some hepatic excretion also occurs, but is not clinically significant. Hirudin should not be used in patients with renal failure, because no specific antidote exists should overdose occur.
Bivalirudin.
Bivalirudin is a recombinant protein based on hirudin. It is a 20–amino acid peptide that interacts with the active site of thrombin.34 It has a short half-life of 25 minutes. In contrast to hirudin, renal excretion is not the major route of excretion.36
Argatroban.
Argatroban is a synthetic, small-molecule arginine derivative that interacts only with the active site of thrombin.34,36 It is metabolized by the liver, with a half-life of 45 minutes. Dose reduction may be necessary in patients with liver dysfunction. The anticoagulant effect can be monitored with the aPTT or ACT.
Dabigatran
Structure and Mechanism of Action.
The oral direct thrombin inhibitor most recently approved by the U.S. Food and Drug Administration (FDA) is dabigatran etexilate. It is a synthetic, nonpeptide, direct thrombin inhibitor which inhibits both circulating and clot-bound thrombin and lowers thrombin-stimulated platelet aggregation.37 It is rapidly absorbed from the gut. Peak concentrations occur approximately 1 hour after oral administration, but slows to approximately 3 hours when taken with a high-fat meal. The drug is mainly renally excreted and has a half-life of 12 to 17 hours. Renal impairment can increase serum concentrations of dabigatran. The anticoagulation effect is quite stable40 and monitoring is not required, which makes this drug a potential alternative to warfarin. Unlike warfarin, however, there is no effective antidote, although it is dialyzable. Coadministration of dabigatran with other P-glycoprotein transporter substrates, such as rifampin decreases serum concentrations of dabigatran and may lower its effectiveness. P-glycoprotein inhibitors such as ketoconazole may increase serum concentrations.41
Clinical Use.
Dabigatran has been initially approved by the FDA for prevention of thromboembolic stroke in patients with nonvalvular atrial fibrillation. The Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial studied 18,113 patients with atrial fibrillation considered high risk for stroke. Over a median 2-year of follow-up, they found a decrease in the rate per year of stroke or systemic embolism with dabigatran compared with warfarin (1.54% versus 1.71%; p < 0.05).42 Since 2008, dabigatran has been used in Canada for prevention of thromboembolism in orthopedic surgery patients undergoing knee or hip replacement. In two studies, dabigatran was noted to be as effective as enoxaparin in preventing venous thromboembolism (VTE) after surgery, with no significant hepatotoxicity and a similar incidence of major bleeding. Administration of both 150 and 220 mg dabigatran was as safe and effective as enoxaparin (40 mg) in preventing VTE and all-cause mortality in both the RE-NOVATE and RE-MODEL trials.41,43 In the RE-NOVATE trial, although 24% of patients studied were not available for assessment for the primary efficacy outcome, in those that could be assessed, results showed similar efficacy with both doses of dabigatran and enoxaparin. However, in another trial evaluating VTE, the RE-MOBILIZE study, dabigatran failed to show equivalence to a higher dose of enoxaparin (60 mg).44 For prevention of VTE, clinical investigation continues for dabigatran.
Factor X Inhibitors
Fondaparinux
Fondaparinux is a pentasaccharide that activates antithrombin III in a manner similar to heparin, leading to inactivation of factor X.47 It differs in one major respect from heparin, in that the pentasaccharide is unable to mediate the inactivation of thrombin by antithrombin III.47 It is administered subcutaneously and does not require laboratory monitoring. It is renally excreted and is contraindicated in patients with renal failure. Fondaparinux has been evaluated in multiple trials for preventing deep venous thrombosis in patients undergoing major hip fracture and knee replacement surgery.48–51 It also has been evaluated in the treatment of deep venous thrombosis52 and acute coronary syndromes.53
Rivaroxaban
Clinical Use.
Rivaroxaban received FDA approval in 2011 for prevention of VTE in hip and knee arthroplasty. A series of phase III trials of rivaroxaban evaluated almost 10,000 patients undergoing elective knee or hip arthroplasty (RECORD 1, 2, and 3 trials).54–56 Results from these studies showed the superior efficacy of rivaroxaban compared with enoxaparin. In a study of 2531 patients undergoing knee arthroplasty, the composite primary end point (any deep vein thrombosis [DVT], nonfatal pulmonary embolism [PE], or death from any cause) after 10 to 14 days of therapy was noted in 9.6% of patients taking rivaroxaban compared with 18.9% of patients receiving enoxaparin; major bleeding occurred in 0.6% and 0.5% of patients, respectively.55 In 3153 hip arthroplasty patients, after 36 days of therapy the same primary endpoint was noted in 1.1% of patients on rivaroxaban and in 3.7% of patients taking enoxaparin. Major bleeding occurred in 0.3% of patients with rivaroxaban and in 0.1% with enoxaparin (p > 0.05).54
In all three trials, rivaroxaban and enoxaparin had comparable safety profiles and low rates of major bleeding. A pooled analysis from REgulation and Congulation in ORthopedic Surgery to Frequent Deep Venous Thrombosis and Pulmonary Embolism (RECORD) 1, 2, and 3 showed that compared with enoxaparin, rivaroxaban decreases the composite of symptomatic VTE and all-cause mortality after elective total hip or knee arthroplasty, with a small increase in bleeding, no evidence of hepatotoxicity, and fewer serious adverse events.57 Furthermore, rivaroxaban has been shown to have a significantly lower rate of recurrent DVT than warfarin-treated patients in the EINSTEIN DVT study.58 And rivaroxaban has been found to be as effective as warfarin for preventing stroke in patients with atrial fibrillation in the Rivaroxaban-Once daily, oral, direct factor Xa inhibition Compared with vitamin K antagonism for prevention of stroke and Embolism Trial in Atrial Fibrillation (ROCKET-AF) study. Throughout all these studies, rivaroxaban has maintained a slightly better safety profile.59
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


