(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
The majority of the cellular energy is produced in the form of ATP by the oxidative phosphorylation (OXPHOS) pathway, through the oxidation of organic substrates, mainly carbohydrates and fatty acids. Carbohydrates initially undergo conversion into pyruvate via glycolysis. Pyruvate then enters mitochondria and transforms to acetyl-CoA by the mitochondrial pyruvate dehydrogenase complex for further oxidation in the tricarboxylic acid (TCA) cycle. Fatty acids also translocate to mitochondria for β-oxidation. Analogous to carbohydrate metabolism, the end product of fatty acid β-oxidation is acetyl-CoA. This intermediary metabolite is “burned” to carbon dioxide via cascade of enzymatic reactions, known as the TCA (or Krebs) cycle to produce high-energy reducing equivalents, in the form of NADH and FADH2. Oxidation of NADH and FADH2 is a series of redox reactions catalyzed by components of mitochondrial respiratory electron transport chain (ETC). High-energy electrons, released from oxidized reducing equivalents, flow through the respiratory chain and finally reduce molecular oxygen-generating molecules of water. Energy of electrons is transformed into the electrochemical gradient of protons across mitochondrial inner membrane, which is utilized by ATP synthase to phosphorylate ADP into ATP. Several mitochondrial kinases orchestrate coupling of ATP synthesis (in mitochondria) with ATP utilization (in different cell compartments).
Introduction
Sources of energy (fatty acids (FA), pyruvate, glucose) are converted in mitochondria into reduced NAD (NADH) and FAD (FADH2). Oxidation of them by components of mitochondrial respiratory electron transport chain (ETC) is accompanied by release of electrons, directional movement of the electrons between key ETC carriers, and transferring to the final acceptor, molecular oxygen. Energy released from electrons transferring at specific sites of ETC is used to pump protons across the mitochondrial inner membrane (from the matrix to the intermembrane space) generating thereby electrochemical proton gradient. Backflow of protons is utilized by ATP synthase (a reverse proton pump), an enzyme that phosphorylates ADP.
Initially all carbohydrates undergo conversion into pyruvate via glycolysis in the cytosol. Pyruvate in order to enter mitochondria for further metabolic reactions is transformed to acetyl-CoA by the mitochondrial pyruvate dehydrogenase complex (PDHC). PDHC is a highly regulated enzyme promoting tissue-specific fuel consumption and storage. For instance, PDHC activity is restricted in myocardium under normal conditions when FAs or ketone bodies serve as primary fuel sources. Under stress conditions (e.g., exercise), increase of the PDHC activity correlates with the oxidative use of glycogen and blood glucose.
A major fuel in muscle such as heart and skeletal muscle is FAs. Once entering the myocyte, FA is directed to β-oxidation in mitochondria. Process of β-oxidation is tightly regulated by interactions between the components of so-called carnitine shuttle responsible for entering long-chain (LC) FAs (LCFAs) to the mitochondria. Within mitochondria, LCFA is released to the mitochondrial matrix in the form of the LC acyl-CoA and is oxidized by the enzymes of the β-oxidation spiral pathway. The β-oxidation process consists of four enzymatic steps catalyzed by several enzymes with their own FA chain-length specificity. Main end product of β-oxidation is acetyl-CoA.
Both PDHC-catalyzed reaction and FA β-oxidation process result in the formation of intermediary metabolite acetyl-CoA, which is fed into the tricarboxylic acid (TCA) cycle (or Krebs cycle) to produce high energy reducing equivalents, NADH and FADH2. Very important mitochondrial component, ETC, produces energy by transfer electrons from reducing equivalents to oxygen through four multienzyme complexes (I–IV) thereby creating a proton electrochemical gradient across the mitochondrial inner membrane. This gradient drives the FO–F1 ATP synthase (complex V), which produces ATP by phosphorylation of ADP.
The cellular bioenergetic system for optimal operation requires that energy-rich phosphoryls (mainly ATP) are produced and delivered to energy-consuming sites (myofibrils in the heart) at the rate corresponding to the ATPase velocity. Also products of ATP hydrolysis (ADP, Pi, H+) must be removed in order to avoid kinetic and thermodynamic difficulties. There are several intracellular enzymatic networks, catalyzed by creatine kinase, adenylate kinase, and nucleoside diphosphate kinase, that support high-energy phosphoryl transfer and signal communication between ATP-generating and ATP-consuming processes. For instance, mitochondrial creatine kinase catalyzes the transfer of the high-energy phosphate bond in ATP to creatine to form phosphocreatine (PCr) and ADP. PCr, in contrast to ATP, is able to rapidly diffuse from the mitochondria to the myofibrils, where myofibrillar creatine kinase catalyzes the reformation of ATP from PCr. Creatine kinase system acts as an energy buffer to keep ATP at normal levels.
Pyruvate Dehydrogenase Complex
All carbohydrate sources of calorie intake are converted into pyruvate in the cytosol via glycolysis. In addition, about one-half of carbon skeletons of gluconeogenic amino acids also convert into pyruvate. Pyruvate enters mitochondrion through the transporter (PyT) located in the mitochondrial inner membrane. 41.9-kDa yeast pyruvate carrier has been shown to belong to mitochondrial carrier family [1]. The mammalian PyT can be inhibited very specifically and effectively by thiazolidine compounds [2], but its molecular entity is yet to be identified. Within mitochondria pyruvate serves as a substrate for pyruvate dehydrogenase complex localized in mitochondrial matrix. Approximately 50% of daily calorie intake passes through PDHC. PDHC catalyzes irreversible oxidative decarboxylation of pyruvate to acetyl-CoA, thereby linking the glycolysis to the oxidative pathway of the TCA.
Chemical reaction catalyzed by PDHC proceeds via the sequential steps mediated by protein components of PDHC, pyruvate dehydrogenase (E1), dihydrolipoyl acetyltransferase (E2), and dihydrolipoyl dehydrogenase (E3) (Fig. 5.1). E1 performs the first two reactions: a decarboxylation of pyruvate and a reductive acetylation of lipoic acid. Mammalian E1 is a tetramer, composed of two α- and two β-subunits, with two catalytic sites (each formed at the interface of α- and β-subunits), two coenzyme molecules (thiamine pyrophosphate, TPP), and two Mg2+ as cofactors. First, TPP in the active center of E1 decarboxylates pyruvate, and acetyl portion covalently attaches to TPP. The carbon dioxide produced by this reaction is nonpolar and small and can diffuse out of the mitochondria and out of the cell. Then E1 catalyzes reductive acetylation of lipoic acid: transfer of acetyl from TPP to lipoic acid (cofactor covalently bound to E2- and E3-binding protein, E3BP). The latter reaction is normally rate limiting in mammalian PDHC. Three isoforms of E1 α-subunit, PDHA1, PDHA2, and PDHB, are encoded by three distinct nuclear genes.
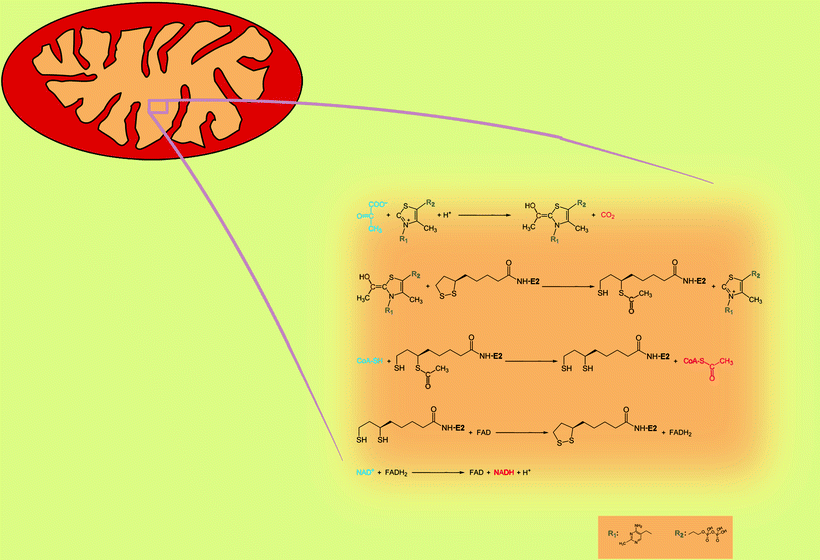
Fig. 5.1
The reactions catalyzed by the pyruvate dehydrogenase complex. Blue color, substrates; red color, products
E2 is the second of the three enzyme components participating in the conversion of pyruvate into acetyl-CoA. E2 catalyzes transfer of acetyl group from lipoic acid to CoA. Thus, E2 produces the end product of pyruvate decarboxylation, acetyl-CoA, and reduced form of lipoic acid, dihydrolipoamide. Acetyl-CoA released from the E2 subsequently enters the TCA cycle.
E3 is a flavoprotein containing flavin adenine dinucleotide (FAD), which regenerates the cofactor on E2 by conversion of dihydrolipoamide into lipoamide. Besides regeneration of lipoic acid, E3 produces an essential side product, NADH. Interestingly, dihydrolipoyl dehydrogenase is also a component of the α-ketoglutarate dehydrogenase complex, the branched-chain α-keto acid dehydrogenase complex, and the mitochondrial glycine cleavage system.
All enzymes participating in the conversion of pyruvate into acetyl-CoA are organized into multimeric complex, PDHC. The central PDHC core contains 60 E2 molecules arranged into an icosahedron. Recently, it has been suggested that 12 molecules of E2 in bovine PDHC core are replaced by E3BP [3]. In eukaryotes, E1 tetramer (up to 20–30) specifically binds to E2 [4], while up to 12 E3 dimers associate with E3BP [3].
In addition to enzymes converting pyruvate to acetyl-CoA, PDHC contains also pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) which play an important role in the regulation of PDHC activity via reversible phosphorylation of three sites on the E1. Four PDK isoforms, PDK1, PDK2, PDK3, and PDK4, comprise a family of serine kinases unrelated to other Ser/Thr kinases [5, 6]. The PDKs are dimers [7, 8] and bind to E2 lipoyl domains by providing enhanced access to the E1 substrate [9] which becomes inactive in phosphorylated state.
Two isoforms of PDP catalytic subunits, PDP1 and PDP2, belong to the 2C class of protein phosphatases [10, 11]. PDP2 is expressed in fat-synthesizing tissues [10] and is activated by insulin via allosteric activation [12, 13] and PKCδ-dependent phosphorylation [14]. Insulin enhances activity of PDP2 by lowering of the K m for Mg2+ [15, 16]. Besides 52-kDa catalytic subunit (PDP1c), PDP1 also contains 95.6-kDa regulatory subunit (PDP1r) [17]. Importantly, PDP1 is found in Ca2+-sensitive tissues and is activated by micromolar Ca2+ [18]. PDP binds to E2 lipoyl domains.
Increased ratios of NADH/NAD+ and acetyl-CoA/CoA observed under increased use of FAs and ketone bodies activate PDK with subsequent inactivation of PDHC [19, 20]. On the other hand, increases in glucose transport or glycogen breakdown elevate pyruvate and ADP, which synergistically inhibit the PDK activity leading to higher activity of PDHC [9, 21]. (More about regulation of PDK itself and PDK/PDP-dependent regulation of PDHC, see Chap. 6.)
Fatty Acid Oxidation (β-Oxidation Spiral)
The major source of energy for skeletal muscle and the heart is FAs. They become incorporated into cellular bioenergetics via mitochondrial β-oxidation pathway (FAO) with subsequent entering TCA cycle. FAO also takes place in the liver during increased physical activity, under the conditions of prolonged fasting, during illness. In addition, hepatic FAO is a component of the synthesis of ketone bodies which fuel the brain when blood glucose levels are low. The FAO pathway is comprised of about 25 enzymes and transport proteins. Deficiencies in many of them have been demonstrated to cause disease in humans.
The primary sources of FAs are triglycerides from animal fats and plant oils. Digestion of triglycerides by lipases releases free FAs which penetrate through the intestinal wall into the lymph and bloodstream and can be either β-oxidized by tissues requiring energy or stored in adipose tissue in the form of lipids. For the generation of metabolic energy from adipose lipids, they undergo lipolysis, and free FAs release into the bloodstream. Plasma FAs are actively transported into the cell, where medium- and short-chain FAs are thought to directly enter the mitochondrial matrix where β-oxidation-related enzymes are located [22]. Long-chain and very-long-chain FAs have to be esterified to coenzyme A by acyl-CoA synthase on the mitochondrial outer membrane (MOM) with consumption of energy from ATP and then translocated to the mitochondrial matrix.
Translocation of long-chain and very-long-chain fatty acyl moieties across the mitochondrial inner membrane (MIM) is mediated by the carnitine shuttle mechanism [23] (Fig. 5.2). First, acyl-CoA is conjugated to carnitine by carnitine acyltransferase I (carnitine palmitoyltransferase I, CPT-I) tightly bound to the MOM. Then acylcarnitine is shuttled inside through the intermembrane space by a carnitine–acylcarnitine translocase (CAT). Finally, carnitine acyltransferase II (carnitine palmitoyltransferase II, CPT-II) loosely associated with the inner side of the MIM converts acylcarnitine to acyl-CoA. The liberated carnitine returns to the cytosol, while acyl-CoA enters FAO.
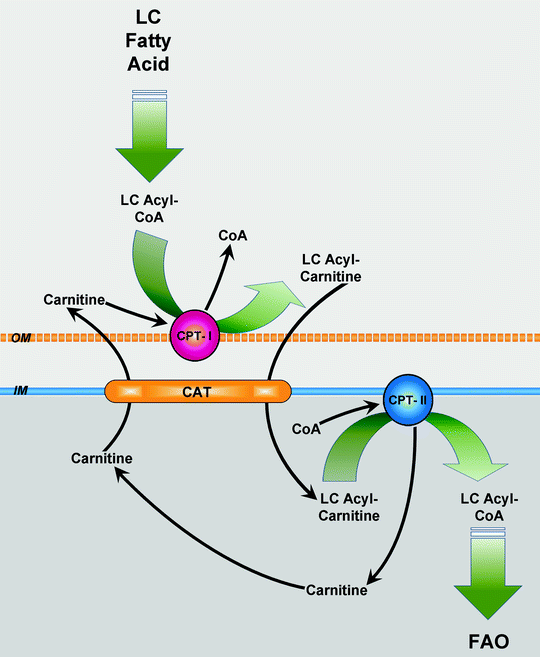
Fig. 5.2
Carnitine shuttle. Upon its uptake, long-chain (LC) fatty acid is converted to LC fatty acyl-coenzyme A (LC acyl-CoA) by acyl-CoA synthetase. LC acyl-CoA is imported into mitochondria via the carnitine shuttle, consisting of carnitine palmitoyltransferase (CPT)-I, carnitine–fatty acylcarnitine translocase (CAT), and CPT-II. Subsequently, the transporter CAT exchanges one molecule of long-chain fatty acylcarnitine (LC acylcarnitine) for one molecule of carnitine. In mitochondrial matrix, LC acyl-CoA is used for fatty acid β-oxidation (FAO). Other abbreviations: IM mitochondrial inner membrane, OM mitochondrial outer membrane
There are three tissue-specific isoforms of CPT-I in humans which are products of different genes: hepatic CPT-IA, muscle CPT-IB, and brain CPT-IC [23, 24]. CPT-IA is the main isoform in adult liver, lung, spleen, intestine, ovary, and pancreas. It is also expressed in the heart of the late fetal and neonatal rat, but is replaced by CPT-IB during later development [25]. CPT-IB predominates in skeletal muscle, adult heart, adipocytes, and testes. All three isoforms of CPT-I are expressed in different regions of brain [26]. The CPT-I-catalyzed reaction is the rate-determining step in mitochondrial FA β-oxidation. CPT-I undergoes allosteric regulation (inhibition) by malonyl-CoA, an intermediate in fatty acid biosynthesis, to prevent futile cycling between β-oxidation and fatty acid synthesis. Regulation of CPT-I by malonyl-CoA is discussed in Chap. 6.
CAT catalyzes facilitated diffusion of different acylcarnitine species to enter the mitochondrial matrix in exchange with carnitine from the mitochondrial matrix. It contains two membrane-spanning α-helices joined by hydrophobic regions, similarly to the other mitochondrial carriers. CPT-II gene encodes a protein 658 amino acids long present in all tissues.
Once in the mitochondrion, FA enters a spiral process of β-oxidation. In each step of the spiral, four chemical reactions catalyzed by two to four nuclear-encoded enzymes take place. At the end of each step, a two-carbon fragment is removed from the acyl moiety in the form of acetyl-CoA, and the remaining acyl-CoA, two carbons shorter, enters the next step (Fig. 5.3).
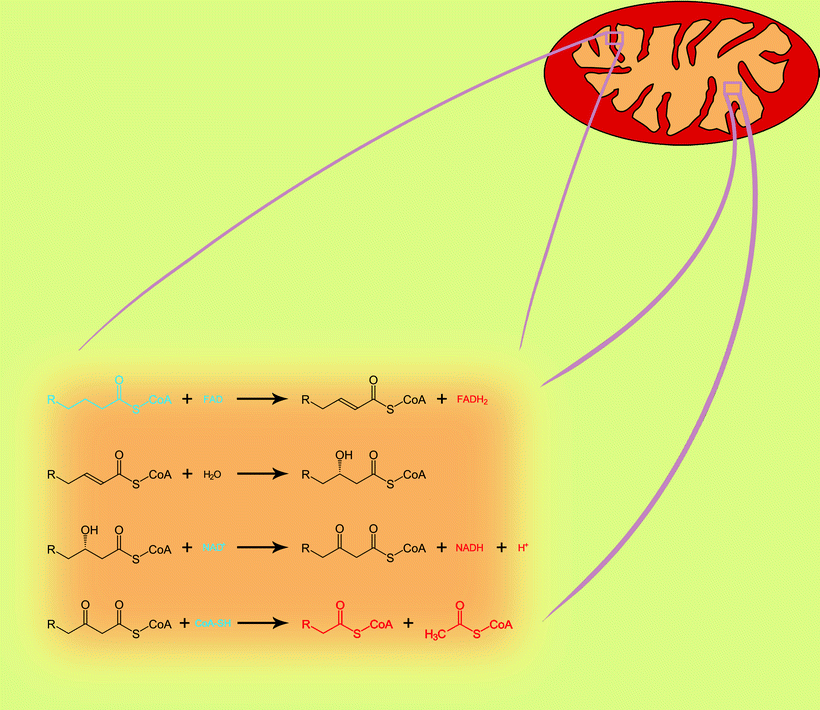
Fig. 5.3
The four reactions of mitochondrial fatty acid β-oxidation pathway. One step of spiral process is shown. Blue color, substrates; red color, products
The first phase of the acyl-CoA β-oxidation is dehydrogenization catalyzed by a family of FAD-dependent acyl-CoA dehydrogenases that exhibit different substrate specificities. Thus, very-long-chain acyl-CoAs are substrates for very-long-chain acyl-CoA dehydrogenase (VLCAD), whereas long-chain acyl-CoAs are consumed by long-chain acyl-CoA dehydrogenase (LCAD). Similarly, medium-chain acyl-CoA dehydrogenase (MCAD) and short-chain acyl-CoA dehydrogenase (SCAD) are known that utilize medium-chain acyl-CoAs and short-chain acyl-CoAs, respectively. FAD is required for the enzyme to bind to its appropriate substrate, and it oxidizes fatty acyl (Fig. 5.4). Resulting products of dehydrogenization process are high-energy compounds, FADH2, and 2,3-enoyl-CoA.
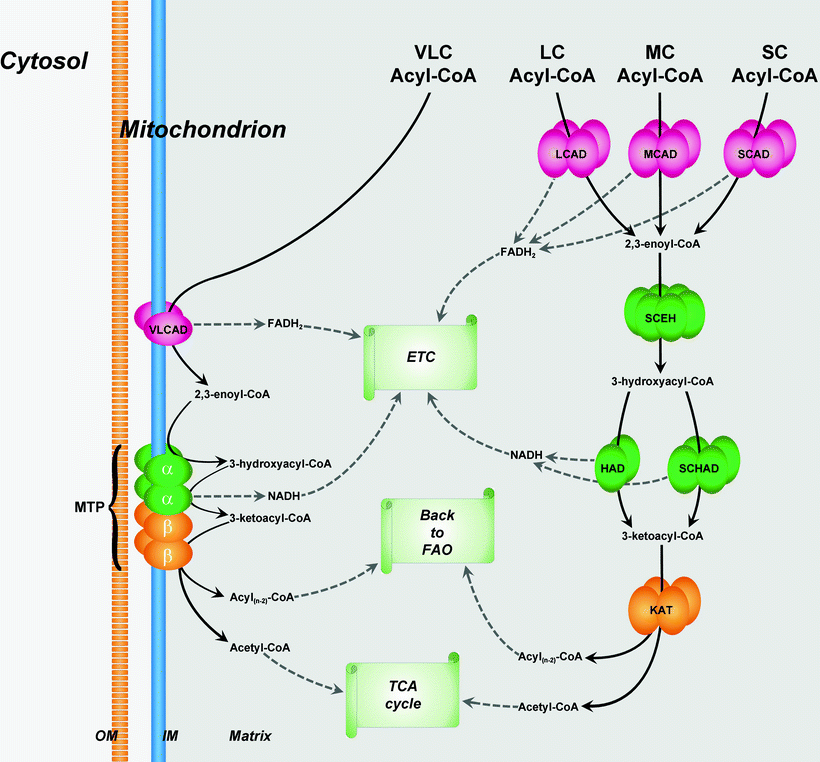
Fig. 5.4
Organization of fatty acid β-oxidation enzymes in mitochondria. β-oxidation pathway (FAO) of very-long-chain (VLC) fatty acids is localized in the inner membrane (IM) of mitochondria, whereas β-oxidations of long-chain (LC), medium-chain (MC), and short-chain (SC) fatty acids take place in the mitochondrial matrix. See text for details. Abbreviations: CoA coenzyme A, ETC electron transport chain, HAD 3-hydroxyacyl-CoA dehydrogenase, KAT 3-ketoacyl-CoA thiolase, LCAD LC acyl-CoA dehydrogenase, MCAD MC acyl-CoA dehydrogenase, MTP mitochondrial trifunctional protein, OM outer membrane of mitochondria, SCAD SC acyl-CoA dehydrogenase, SCEH SC 2,3-enoyl-CoA hydratase, SCHAD SC 3-hydroxyacyl-CoA dehydrogenase, TCA cycle tricarboxylic acid cycle, and VLCAD VLC acyl-CoA dehydrogenase. Metabolic conversions related to FAO are shown by black solid arrows
Enoyl derivative of acyl-CoA is hydrated then by enoyl-CoA hydratase. Two enzymes are described with different substrate specificity: long-chain 2,3-enoyl-CoA hydratase (LCEH) and a short-chain 2,3-enoyl-CoA hydratase (SCEH). They hydrate the 2,3-enoyl-CoA across the double bond, with the release of 3-hydroxyacyl-CoA.
Next enzyme, NAD-dependent 3-hydroxyacyl-CoA dehydrogenase, oxidizes the 3-hydroxy position producing a 3-ketoacyl-CoA. Oxidation of 3-hydroxyacyl-CoA is accompanied by the production of high-energy compound, NADH. A long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) and 3-hydroxyacyl-CoA dehydrogenase (HAD) oxidize long-chain and medium-chain 3-hydroxyacyl-CoAs, respectively. Third mitochondrial dehydrogenase, called short-chain 3-hydroxyacyl-CoA dehydrogenase (SCHAD, also known as 17β-hydroxysteroid dehydrogenase type 10), acts on a wide spectrum of substrates, including steroids, cholic acids, and FAs.
The last phase in every cycle of the FAO spiral is mediated by 3-ketoacyl-CoA thiolase. Three different 3-ketoacyl-CoA thiolases include a long-chain 3-ketoacyl-CoA thiolase (LKAT), medium-chain 3-ketoacyl-CoA thiolase (MKAT), and short-chain 3-ketoacyl-CoA thiolase (SKAT). They all shorten acyl-CoA by two carbons by cleaving off and transferring acetyl group to the fresh CoA. The shortened acyl-CoA can then reenter the FAO spiral until the FA is completely broken down into a two- or three-carbon species. The produced acetyl-CoA can be used as a substrate for TCA cycle (see below).
VLCAD is a homodimer located on the MIM. It is a main rate-limiting enzyme of FA β-oxidation, whereas role of LCAD in energy production is not essential. Enzymes, mediating three last reactions of long-chain FA oxidation, are assembled in the mitochondrial trifunctional protein (MTP) enzyme complex also located on the MIM [27]. MTP is a heterotrimeric protein that consists of four α-subunits and four β-subunits. The amino-terminal domain of the α-subunit contains the LCEH enzymatic activity, whereas the LCHAD enzymatic activity resides in the carboxy-terminal domain. The β-subunit has the LKAT activity (Fig. 5.4).
Enzymes involved in the degradation of medium- and short-chain FAs (MCAD, SCAD, SCEH, HAD, SCHAD, SKAT) are present in the mitochondrial matrix [27]. MCAD is a homotetramer containing four FAD molecules and four acyl-CoA-binding active sites. HAD is a homodimer (two 33-kDa subunits), whereas SCHAD is a homotetramer (Fig. 5.4).
VLCAD and MCAD are among key enzymes involved in FAO, and their deficiency is associated with cardiomyopathy, muscle weakness, and hepatic dysfunction [28–30]. Patients with defects in the MTP develop cardiomyopathy, skeletal myopathy, and neuropathy and demonstrate a serious liver phenotype [31, 32]. Malfunctioning of SCHAD is related to the neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, and others [33–35].
Production of Reducing Equivalents: Tricarboxylic Acid Cycle
Tricarboxylic acid (TCA) cycle (also known as the citric acid cycle, or Krebs cycle) is one of the most important mitochondrial processes to generate reducing equivalents and to supply the respiratory chain with them (in the form of NADH and FADH2) for ATP synthesis (Fig. 5.5). In addition, TCA cycle provides NADPH for the reduction of H2O2 and glutathione disulfides in mitochondria and supplies intermediates essential for FA and protein synthesis. The TCA cycle is part of a metabolic pathway converting carbohydrates, FAs, and proteins into CO2 and water to produce a bioenergy in the form of ATP.
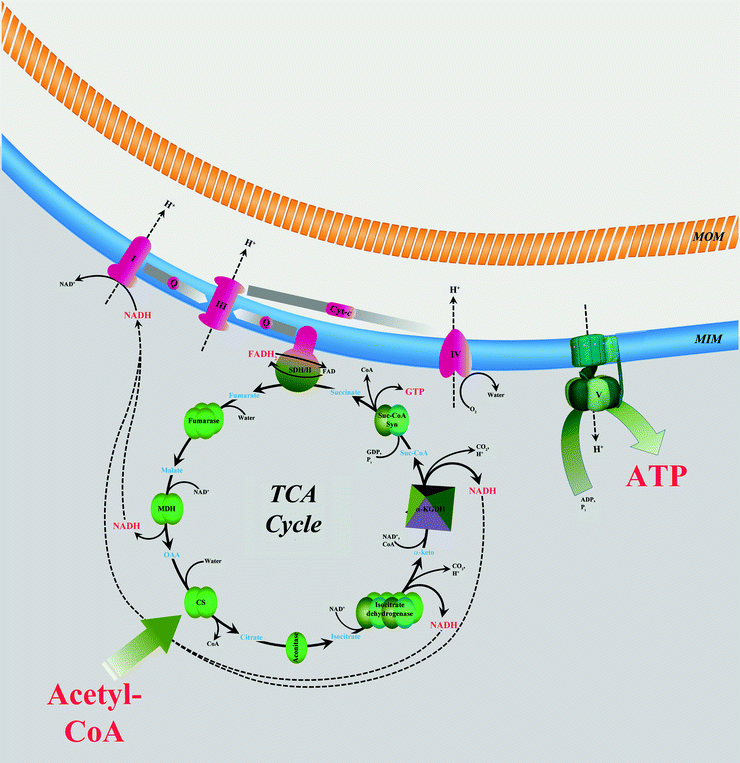
Fig. 5.5
Interaction between tricarboxylic acid cycle and oxidative phosphorylation. Two-carbon-organic fuel enters the tricarboxylic acid (TCA) cycle in the form of acetyl-CoA. NADH and FADH2 generated in the TCA cycle are oxidized by mitochondrial electron transport chain (complexes I–IV) located in the mitochondrial inner membrane (MIM). Energy released is used to generate potential energy in the form of a proton gradient across MIM. This energy powers synthesis of ATP catalyzed by ATP synthase (complex V). See text for details. Abbreviations: I NADH–ubiquinone oxidoreductase, III ubiquinol–cytochrome c oxidoreductase, IV cytochrome c oxidase, CoA coenzyme A, CS citrate synthase, cyt-c cytochrome c, α-keto α-ketoglutarate, α-KGDH α-ketoglutarate dehydrogenase, MDH malate dehydrogenase, MOM mitochondrial outer membrane, OAA oxaloacetic acid, Q ubiquinol/ubiquinone, SDH/II succinate dehydrogenase/succinate–ubiquinone oxidoreductase, Suc-CoA succinyl-CoA, and Suc-CoA Syn Suc-CoA synthetase. Metabolic conversions are shown by black solid arrows. NADH and H+ fluxes are shown by black dashed arrows
Citrate Synthase
Сitrate synthase catalyzes the first step in the TCA cycle: transfer of the acetate group of acetyl-CoA to oxaloacetate, producing citrate. The enzyme is a homodimer composed of two 50-kDa subunits. Each subunit contains two domains separated by a deep cleft forming the active site of the enzyme. A number of substances, including succinyl-coenzyme A and adenine nucleotides, are inhibitors of citrate synthase.
Aconitase
Aconitase catalyzes the reversible isomerization of citrate and isocitrate via cis-aconitate. This process involves four stereospecific reactions: the dehydration of citrate or isocitrate to cis-aconitate and the rehydration of cis-aconitate to isocitrate or citrate. Mitochondrial aconitase is a monomeric 83-kDa protein containing labile [4Fe–4S]2+ cluster.
The aconitase plays a role in oxidative stress: it shows a high sensitivity to H2O2, being completely inactivated at very low H2O2 concentrations (less than 50 μM) and other ROS [36, 37], either due to interaction with the iron–sulfur cluster [38, 39] or indirectly via interaction between the enzyme and a membrane component responsive to peroxide [39]. Inhibition of aconitase could lower the flux through the cycle due to accumulation of citrate and a consequent inhibition of citrate synthase. Yarian et al. [40] have demonstrated that the activity of aconitase is altered in rats during the aging process. They suggested that an age-related decrease in aconitase activity may be associated with an age-related increase in the prooxidant state in mitochondria and is likely to contribute to a decline in the overall efficiency of mitochondrial bioenergetics.
Isocitrate Dehydrogenase
Mammalian NAD isocitrate dehydrogenase exists primarily as an octamer of composition (α2βγ)2. Three different subunits of the enzyme are structurally related and have molecular masses around 40 kDa [41, 42]. Substrate binding and active sites are shared between the subunits [43, 44]. The enzyme plays a controlling role in the formation of NADH for the respiratory chain and shows complex regulatory properties. In particular, the enzyme is inhibited by increasing ATP/ADP and NADH/NAD+ ratios. Calcium ions activate mammalian NAD isocitrate dehydrogenase, and the enzyme is more sensitive to Ca2+ at lower ATP/ADP ratios [45, 46].
Alternatively, isocitrate can be converted to 2-oxoglutarate in mitochondria by NADP isocitrate dehydrogenase [45], and reducing equivalents from produced NADPH can be transferred to NAD+ by nicotinamide transhydrogenase localized in the MIM, to give NADH [47]. NADP isocitrate dehydrogenase shows no allosteric regulation [45]. Normally, it is the regulated NAD+-linked enzyme that participates in the TCA cycle [48].
2-Oxoglutarate Dehydrogenase (α-Ketoglutarate Dehydrogenase)
Similarly to PDHC, 2-oxoglutarate dehydrogenase is a multienzyme complex (OGDHC) performing oxidative decarboxylation of 2-oxoglutarate (OG). Chemical reaction catalyzed by OGDHC proceeds via the sequential steps catalyzed by protein components of OGDHC, 2-oxoglutarate dehydrogenase (E1), dihydrolipoyl succinyl transferase (E2), and dihydrolipoyl dehydrogenase (E3). E1 performs the first two reactions: a decarboxylation of 2-oxoglutarate and a reductive succinylation of lipoic acid. E1 contains coenzyme molecule (thiamine diphosphate, TDP) and Mg2+ as a cofactor. First, TDP in the active center of E1 decarboxylates 2-oxoglutarate, and hydroxybutyrate fragment covalently attaches to TDP. The carbon dioxide produced by this reaction is nonpolar and small and can diffuse out of the mitochondria and out of the cell. Then E1 catalyzes reductive succinylation of lipoic acid: transfer of succinyl from TDP to lipoic acid (cofactor covalently bound to E2). E1 has the lowest catalytic activity among the components and as such limits the rate of the overall OGDHC-catalyzed process [49]. As with pyruvate dehydrogenase, it is likely that the step catalyzed by E1 normally regulates the overall OGDHC activity [50].
E2 is the second of the three enzyme components participating in the conversion of 2-oxoglutarate into succinyl-CoA. E2 catalyzes transfer of succinyl group from lipoic acid to CoA. Thus, E2 produces the end product of 2-oxoglutarate decarboxylation, succinyl-CoA, and reduced form of lipoic acid, dihydrolipoamide. Succinyl-CoA released from the E2, continues further degradation within the TCA cycle.
E3 is the same flavoprotein as the one contained in the PDHC (see above) and serves same functions—regeneration of the cofactor on E2 (by conversion of dihydrolipoamide into lipoamide) and production of NADH.
All enzymes, participating in the conversion of 2-oxoglutarate into succinyl-CoA, are organized into multimeric complex, OGDHC. The central OGDHC hollow is made of predominantly 24 E2 molecules, to which are attached dimeric E1 and E3 subunits (the latter attachment may be also via binding to E1 subunits) [51, 52]. Each OGDHC probably contains six E1 dimers.
As with pyruvate dehydrogenase, the 2-oxoglutarate dehydrogenase is end product inhibited by increases in the succinyl-CoA/CoA, NADH/NAD+, and ATP/ADP ratios. However, unlike pyruvate dehydrogenase, the enzyme is not regulated by reversible phosphorylation. 2-oxoglutarate dehydrogenase is directly activated by Ca2+, and decreases in ATP/ADP ratio markedly increase the sensitivity of the enzyme to calcium ions [46, 50]. Unlike isocitrate dehydrogenase, 2-oxoglutarate dehydrogenase is an order of magnitude more sensitive to activation by Ca2+. OGDH has been found to be inhibited in cardiac mitochondria under myocardial ischemia and reperfusion [53]. This inactivation involves ROS-dependent oxidative damage of lipoic acid essential for the catalytic activity of E2.
It seems worth to note that OGDHC also catalyzes several side reactions of potential biological significance. Under the conditions of excessive accumulation of the dihydrolipoamide intermediate, OGDHC catalyzes oxidation of the latter by molecular oxygen with the formation of the ROS (superoxide anion radical) and thiyl radical of the E2-bound lipoamide [54]. OGDHC-dependent ROS may contribute to cellular signaling and/or damage. Another biologically relevant side reaction of OGDHC is the carboligase activity of its first TDP-dependent E1 component, which appears to be involved in glyoxylate scavenging [55].
Succinate Dehydrogenase
Succinate dehydrogenase (SDH) is a unique enzyme participating in both the TCA cycle and respiratory electron transfer chain, OXPHOS (Fig. 5.5). In this section, we focus on the functioning and the role of SDH in the citric acid cycle, whereas involvement of it in the ETC will be described later.
Within the citric acid cycle, SDH oxidizes succinate to fumarate. Eukaryotic enzyme consists of four subunits. For instance, the porcine cardiac SDH consists of a hydrophilic head that protrudes into the mitochondrial matrix compartment and a hydrophobic tail that is embedded in the MIM [56, 57]. The head consists of two subunits (SdhA, SdhB in mammals) forming the catalytic core. SdhA and SdhB subunits contain the redox cofactors that participate in ETC. 70-kDa SdhA contains the covalently bound FAD cofactor and the binding site for succinate. 27-kDa SdhB contains the three Fe–S centers that mediate electron transfer to ubiquinone [57, 58]. SdhB also forms the interface between the catalytic domain and the membrane domain of the complex (15-kDa SdhC and 12–13-kDa SdhD subunits with heme b binding in mammals).
Oxidation of succinate in the active center of SdhA subunit involves the two-electron-dependent reduction of FAD. Then electrons from FADH2 travel via Fe–S centers of SdhB subunit to the ubiquinone (coenzyme Q, CoQ) bound to QP and QD sites formed by SdhC and SdhD subunits and reduce it to ubiquinol. Thus, SDH links TCA cycle (succinate oxidation) to ETC (ubiquinol formation).
The catalytic activity of SDH is modulated by posttranslational modifications (phosphorylation, acetylation) and by active site inhibition. In mouse SdhA, reversible acetylation at Lys residues was shown to attenuate catalytic activity of SdhA [59]. The level of SdhA acetylation is controlled by deacetylase SIRT3. In mammalian cells the SdhA subunit is phosphorylated and this modification, like acetylation, decreases enzymatic activity [60]. SdhA is also a substrate of the tyrosine kinase Fgr in vitro, although the physiological significance of this modification is not known [61]. SdhA activity is markedly decreased in the post-ischemic myocardium, and this loss of activity is associated with another covalent modification—deglutathionylation of Cys-90. It has been suggested that deglutathionylation facilitates sensitivity of the 70-kDa SdhA subunit to oxidative stress induced by ROS during post-ischemic reperfusion of myocardium [62]. SDH catalytic activity is modulated by TCA-cycle intermediates including oxaloacetate, which is a potent inhibitor. Succinate and ATP facilitate the dissociation of oxaloacetate from SDH thereby activating the enzyme. The oxaloacetate-dependent inhibition contributes to the modulation of SDH activity by the metabolic status of mitochondria [63].
Mutations in SdhA and SdhB impair SDH activity and cause diseases. For example, mutations in SdhA are associated with several types of neurodegenerative disorder in humans (subacute necrotizing encephalomyelopathy, SNEM, also known as Leigh syndrome) [64–67] and with symptoms of late-onset myopathy [68]. It has been speculated that the mutation could alter SdhA’s conformation or redox state which might increase sensitivity of SdhA to the inhibitory effect of oxaloacetate [64]. Mutations in SdhB are associated with paragangliomas 4 (PGL4s)—neuroendocrine tumors that can occur in cells of the neural crest commonly located adjacent to the carotid body, as well as in the adrenal glands (there they are called pheochromocytomas, PCCs) [69–71]. Mechanism of tumor formation may include accumulation of succinate due to dysfunction of SDH [72], succinate-dependent inhibition of cytosolic prolyl hydroxylase (PHD), and the accumulation of hypoxia-inducible factor (HIF) by preventing its degradation and resulting in enhancement of tumorigenesis through the resistance to apoptotic signals [73]. Also the increase in HIF can lead to the stimulation of angiogenesis that will feed and supply the tumor [74].
Fumarase
The active form of fumarate hydratase (FH) is a homotetramer in which three of the four subunits form the enzymatic active site for the reversible conversion of fumarate to malate. Dysfunction of FH leads to the accumulation of fumarate in the mitochondria [72]. Similar to succinate, increased fumarate levels cause in the cytosol series of events that enhance tumorigenesis [72–74]. In the case of FH deficiency, accumulation of intramitochondrial fumarate has been shown to slow down the SDH reaction in skeletal muscle [75].
Malate Dehydrogenase
This enzyme catalyzes the conversion of malate into oxaloacetate. Important malate dehydrogenase (MDH)-catalyzed co-reaction is the production of high-energy equivalent, NADH, which serves as a fuel for mitochondrial OXPHOS. There are two main isoforms of MDH in eukaryotic cells, soluble (MDH1) and mitochondrial (MDH2), both being homodimers of 30–35-kDa subunits. MDH contains two distinct domains, the amino-terminal NAD-binding domain and the carboxy-terminal domain with substrate binding site. The active site of the enzyme is in a cleft between two domains [76]. Mitochondrial MDH2 is highly regulated by TCA-cycle products: high malate concentrations stimulate activity of MDH2, whereas high oxaloacetate concentrations inhibit the enzyme. In addition, citrate can allosterically inhibit MDH2-dependent oxidation of malate when levels of malate and NAD+ are low and stimulate MDH2 in the presence of high levels of malate and NAD+ [77, 78]. According to Mullinax et al. [78], all three effectors bind to the same putative allosteric site on MDH2. Mitochondrial enzyme takes part in the TCA cycle under conditions where the conversion of malate into oxaloacetate is unfavorable (under standard conditions, the MDH-catalyzed reaction tends to form malate). This problem is solved via formation of complex of MDH2 with fumarase and citrate synthase where the removal of oxaloacetate from MDH is facilitated toward citrate synthase, so the TCA cycle is driven toward the oxaloacetate formation [79].
Recently, Wang et al. [80] have reported specific interaction of MDH2 with several subunits of OXPHOS Complex III (bc1 complex, or coenzyme Q: cytochrome c—oxidoreductase) in the heart. Association of MDH is strong (K D is 0.047 μM) and includes interaction with two core subunits (1, 2) and N terminus of subunit 5 (Rieske iron–sulfur protein). Functional consequences of MDH2 binding to Complex III include enhancement of the bc1-activity and activation of MDH2. Interestingly, only reverse reaction (the reduction of oxaloacetate to malate) is stimulated by Complex III. It is suggested that cross talk between Complex III and MDH2 can lead to the increased transport of malate from mitochondria to the cytosol, where it can be converted to oxaloacetate by MDH1 and enter gluconeogenesis.
Soluble isoform of malate dehydrogenase, MDH1, participates in the malate–aspartate shuttle with exchanging reducing equivalents so that malate can pass through the mitochondrial membrane and be transformed into oxaloacetate for further cellular processes. Reducing equivalents are produced in the cytosol during glycolysis in the form of NADH. In order to be utilized in OXPHOS, they have to be transported to mitochondria. However, the MIM is impermeable to NADH, and malate shuttle mechanism exists to carry reducing equivalents across the membrane in the form of malate. MDH1 localized in the mitochondrial intermembrane space oxidizes cytosolic NADH to convert oxoloacetate to malate. Malate is imported into the mitochondrial matrix by malate–oxoglutarate antiporter. In the mitochondrial matrix, malate is converted by mitochondrial MDH into oxaloacetate (see above), during which NAD+ is reduced with two electrons to form NADH and release an H+. As a result, NADH in the mitochondria can be used for OXPHOS to synthesize ATP. MDH1 is highly expressed in cardiac and skeletal muscles where it functions as a part of malate shuttle system for aerobic energy production for muscle contraction [81]. Interestingly, levels of MDH1 increase in the left ventricle of dilated cardiomyopathy patients indicating that MDH1 is involved in the activation of glucose oxidation in hypoxic environment of hypertrophied myocardium [81].
At the end of each TCA cycle, oxaloacetate is regenerated, and the cycle can continue. The NADH generated in the TCA cycle donates electrons in OXPHOS to synthesize ATP. Similarly, FADH2 donates electrons to coenzyme Q, which is an intermediate in the ETC. For each acetyl group entering the TCA cycle in the form of acetyl-CoA, three molecules of NADH, one molecule of ubiquinol, and one molecule of GTP are produced.
Oxidative Phosphorylation
Oxidative phosphorylation (OXPHOS) is the metabolic process that couples oxidation of reducing equivalents derived from nutrient compounds (see previous section, Production of Reducing Equivalents: Tricarboxylic Acid Cycle and ATP synthesis). Oxidation of reducing equivalents, respiration, is a process of sequential transfer of electrons from reducing equivalents (NADH, FADH2) through electron transport chain (ETC) of oxidoreductase reactions, resulting in the reduction of molecular oxygen to water. The ETC includes respiratory complexes I (CI, NADH–ubiquinone oxidoreductase), II (CII, succinate–ubiquinone oxidoreductase), III (CIII, ubiquinol–ferricytochrome c oxidoreductase), and IV (CIV, cytochrome c oxidoreductase, COX). The energy liberated during respiration transforms into the formation of an electrochemical proton gradient across the MIM that is utilized by ATP synthase (respiratory complex V, CV) to produce ATP from ADP and inorganic phosphate (Fig. 5.5). Thus, OXPHOS provides most of the energy used by cell in the form of cellular energy currency, ATP.
ETC components encoded by the nuclear and the mitochondrial genomes are localized in the MIM. Defects in subunits of the ETC result in human mitochondrial diseases.
NADH: Ubiquinone Oxidoreductase (Complex I)
Complex I is the primary entry point of electrons into the ETC: at this step, electrons from NADH are transferred to ubiquinone. NADH oxidation regenerates NAD+ for the TCA cycle and FAO. Potential energy of two electrons transferred from NADH to ubiquinone is used by CI to transport protons across the MIM (four protons are translocated for each two-electron oxidation of NADH). Thus, CI contributes to the development of electrochemical proton gradient.
Net reaction catalyzed by CI:
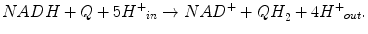
CI is the large multiprotein complex (∼1 MDa) composed of 45 subunits [82, 83]. It is “L” shaped, having two “arms” perpendicular to each other. A hydrophobic “arm” is embedded in the MIM, and a hydrophilic peripheral “arm” protrudes into the mitochondrial matrix [84]. Functionally, there are three modules in CI: the dehydrogenase module composed of the subunits NDUFV2, NDUFV1, and NDUFS1, responsible for the oxidation of NADH to NAD+; the module composed of the subunits NDUFS2, NDUFS3, NDUFS7, and NDUFS8, responsible for transfer of the electron to ubiquinone; and the proton translocation module composed of the seven subunits and located within the membrane “arm” [84–86].
Within the hydrophilic “arm,” one domain is formed by NDUFV1 and NDUFV2. This domain is the place where NADH-binding site and flavin mononucleotide (FMN) are located (both on NDUFV1). Chain of iron–sulfur clusters also starts in this domain. In particular, the iron–sulfur clusters closest to FMN are [2Fe–2S] and [4Fe–4S] non-covalently bound to NDUFV2 and NDUFV1, respectively. This chain continues through another domain formed by [2Fe–2S]- and [4Fe–4S]-containing NDUFS1 and ends in the lower part of hydrophilic “arm” built of NDUFS2, NDUFS3, NDUFS7, and NDUFS8. There are another three [4Fe–4S] iron–sulfur clusters in this part of CI: two of them are non-covalently attached to NDUFS8, and the most distal cluster is attached to NDUFS7. Although the location of ubiquinone-binding site remains unknown, on the basis of site-directed mutagenesis of the NDUFS2 and NDUFS7 subunits, the putative site is usually placed at the interface of NDUFS2 and NDUFS7 subunits in the close proximity to the [4Fe–4S] iron–sulfur cluster on NDUFS7 [87, 88].
Lower part of the hydrophilic “arm” includes the interface with the membrane domain of CI, the hydrophobic “arm.” The latter one is composed of seven highly hydrophobic core subunits—ND1–ND6 and ND4L. Subunits ND4, ND5, and ND2 are related to the Mrp family of Na+/H+ antiporters, and they are often suggested to participate in proton translocation [89].
Prime function of CI upon the binding of NADH is oxidation of NADH by FMN. During oxidation a pair of electrons is transferred from NADH to the flavin. Then they are transferred (one at a time) across the chain of seven iron–sulfur clusters and reunion on a ubiquinone, thus reducing it to ubiquinol. Ubiquinol then dissociates. Ubiquinone (coenzyme Q10, CoQ10) is fat-soluble mobile in mitochondrial membrane molecule. It plays a unique role in the ETC as a carrier of electrons from CI and CII to CIII (see below). At some unknown point, transfer of the electrons from NADH to ubiquinone is coupled to translocation of protons across the membrane.
Under certain conditions an electron of reduced flavin may “escape” from intramolecular traveling through iron–sulfur clusters and hijack a molecule of O2. This results in superoxide production by CI [90]. In addition, [4Fe–4S] iron–sulfur cluster on NDUFS7 subunit as well as semiquinone contributes to ROS production by CI [91].
A number of genetic abnormalities in various subunits and assembly factors of CI lead to the CI deficiency. CI deficiency is the most common cause of mitochondrial disease in both children and adults, and main clinical phenotypes include Leigh syndrome, fatal infantile lactic acidosis (FILA), neonatal cardiomyopathy, leukoencephalopathy, pure myopathy, and combined hepatopathy and tubulopathy [92–95].
Succinate: Ubiquinone Oxidoreductase (Complex II)
Succinate–ubiquinone oxidoreductase (SDH or CII) is a second entry place for electrons from high-energy equivalents formed in mitochondria during the oxidation of pyruvate, FAs, and amino acids. Specifically, the electron transfer pathway from CII-catalyzed oxidated succinate involves initial reduction of a FAD cofactor followed by electron transfer through three Fe–S centers to ubiquinone.
Reactions catalyzed by SDH/CII:
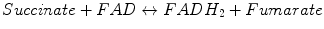
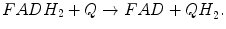
CII is a part of both the TCA cycle and ETC (Fig. 5.5). Eukaryotic CII consists of four subunits and is the only ETC component not pumping protons across the MIM during catalytic cycle. There are two major structural components in the CII: the hydrophilic head formed by SdhA and SdhB, which catalyzes oxidation of succinate during TCA cycle, and a hydrophobic tail mostly embedded in the MIM. We already described the functioning of hydrophilic head (see Production of Reducing Equivalents: Tricarboxylic Acid Cycle), so we concentrate here on the hydrophobic compartment participating in the ETC (to be precise, delivery of electrons from FADH2 to ETC is initiated in the hydrophilic subunit SdhB containing the three Fe–S centers that mediate electron transfer to ubiquinone).
The hydrophobic membrane domain of CII consists of two subunits (SdhC, SdhD). These subunits contain a bound heme b moiety and two ubiquinone-binding sites [57]. The high-affinity ubiquinone site (proximal, QP) is the site where ubiquinone reduction occurs in two stepwise single-electron reactions. QP site stabilizes the partially reduced semiquinone thus preventing dissociation of this potential ROS generator and favoring release of fully reduced ubiquinol [56]. The second site in the hydrophobic domain of CII (distal, QD) has low affinity for ubiquinone. Ubiquinone may undergo reduction at the QD site via electron transfer from the heme, although proof of this is lacking. The significance of the heme moiety and QD site in CII remains unclear.
Ubiquinol–Cytochrome c Oxidoreductase (Complex III, or Cytochrome bc1 Complex, or CIII)
CIII receives electrons from upstream electron transfer complexes (CI and CII) in the form of reduced coenzyme Q10, ubiquinol (QH2). CIII is a dimer embedded in the MIM. In mammals, each monomer is composed of 11 different structural subunits [99]. Three subunits of Complex III contain the metal centers responsible for electron transfer: cytochrome b (subunit MTCYB) contains two b-type heme groups, b L and b H; cytochrome c1 (subunit CYC1) contains heme group c; and Rieske iron–sulfur protein (subunit UQCRFS1, or RISP) contains [2Fe–2S] cluster-binding domain. The function of the other eight subunits remains to be elucidated. CIII catalyzes transfer of electrons from QH2 to cytochrome c (cyt-c). The released energy is used to expel protons from the mitochondrial matrix to the intermembrane space, resulting in the generation of a proton gradient across the MIM. Finally, cytochrome c carries the electrons from CIII to cytochrome c oxidase (CIV).
Three subunits form the intermembrane side of CIII monomer. In particular, it contains the functional domains of cytochrome c1 and the RISP, as well as subunit 8. The transmembrane region of monomer contains membrane-spanning components: cytochrome b; subunits 7, 10, and 11; and the transmembrane helices of the RISP and cytochrome c1. Finally, the side of the CIII which faces mitochondrial matrix is composed of subunits 1, 2, 6, and 9.
According to Q-cycle mechanism, CIII-catalyzed hydroquinone oxidation starts with the binding of QH2 in the intermembrane side of CIII (positive side, QP). Importantly, before QH2 can be oxidized, it must first be deprotonated by releasing two protons into mitochondrial intermembrane space. Each QH2 molecule is a donor of two electrons. One of them is transferred to the 2Fe–2S cluster of RISP. The reduction of the Rieske center increases the affinity of the CIII for semiquinone by several orders of magnitude, stabilizing the semiquinone radical at the QP site. Electron from reduced 2Fe–2S cluster of RISP is transferred to the iron of heme C-containing cytochrome c1. The latter is a 30-kDa subunit of CIII serving as an electron donor to the iron of heme C-containing highly soluble cytochrome c for further transportation to Complex IV. Electron pathway bifurcates during full oxidation of QH2 at the QP site: second electron is accepted by b L heme of cytochrome b (not by RISP). From b L heme this electron travels to b H heme, and finally it becomes accepted by quinone located in the matrix side of CIII (negative side, QN). Complete reduction of quinone at the QN site requires two electrons, so second chain of reactions starting from second QH2 at the QP site completes the whole process. It is important to mention that two protons are recruited by CIII to protonate reduced quinone in the QN site.
Thus, the following processes take place during CIII-dependent electron transfer from ubiquinol to cytochrome c:
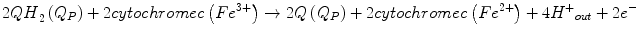
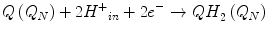
With the resulting net equation:
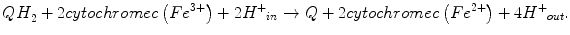
A small fraction of electrons can escape the electron transport chain before reaching CIV and reduce oxygen. Premature electron leakage to oxygen results in the formation of superoxide. Electron leakage occurs mainly at the QP site, when semiquinone reacts with oxygen to form superoxide and other ROS.
Recent evidence demonstrates novel cross talk between CIII and TCA-cycle enzyme, mitochondrial MDH. MDH interacts with CIII subunits 1 and 2 and RISP and can be coprecipitated with bc1 complex. Interaction of bc1 complex with MDH results in an increase of the activities of both the bc1 complex and MDH. Interestingly, CIII-dependent activation of MDH is unidirectional (oxaloacetate → malate). Cross talk between ETC component (CIII) and TCA-cycle enzyme (MDH) might play a regulatory role in mitochondrial bioenergetics [80].
Mutations in the cytochrome b cause CIII deficiency and disorders such as mitochondrial encephalomyopathy, cardiomyopathy, septo-optic dysplasia, and multisystem disorder [100–103]. Similarly, in a murine model of myocarditis (chagasic disease), mice infected with Trypanosoma cruzi demonstrate compromised ATP production in the myocardium because of a decline in cytochrome b expression. Myocardial bioenergetics is disturbed under pathological conditions because of inefficient electron transport for ATP synthesis in mitochondria. Loss in CIII activity can be improved by treatment of infected mice with antioxidant (phenyl-α-tert-butyl nitrone) which restores cytochrome b expression [104]. In addition, a deletion in ubiquinone-binding protein (subunit 7 or QP-C subunit) has been shown to underlie hypoglycemia and liver dysfunction [105].
Cytochrome c Oxidase (CIV or COX)
CIV is the terminal enzyme of the mitochondrial ETC. It catalyzes the electron transfer from reduced cytochrome c to molecular oxygen to form water. The energy released in the reaction of oxygen reduction is conserved in the form of a transmembrane electrochemical gradient of protons across the MIM. Oxidation of every O2 molecule requires four molecules of cytochrome c. The energy released during transfer of electrons from reduced cytochromes c to O2 is used by CIV for net transfer of four protons from the matrix into the MIM, resulting in a proton gradient across the MIM. The overall reaction done by CIV can be described by the following equation:
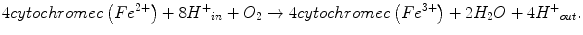
Mammalian CIV is a heteromeric complex composed of 13 different subunits [106]. The structure of this complex has been resolved at present with resolution up to 1.8 Å. The catalytic core of the enzyme is formed by subunits MTCO1 and MTCO2, containing two heme group a moieties (cytochromes a and a 3 ) and the two copper centers (CuA and CuB). Hemes a and copper centers are organized into two binuclear centers (a/CuA and a 3 /CuB) responsible for the electron transfer. Heme a is an essential component of the catalytic centers of CIV found in all eukaryotic and some prokaryotic COX species [107]. More specifically, a 3 /CuB binuclear center and heme a are located on MTCO1, whereas copper center CuA is located on MTCO2 almost on the border with subunit MTCO1. The chemistry of oxygen reduction to water occurs in the a 3 /CuB catalytic site. Subunit MTCO3 is part of the structural core and may play a role in proton pumping.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


