Clinical entity
Clinico-pathologic key points
Main CT scan features
Main Diagnostic step(s)
Therapeutic options
Follicular bronchitis/bronchiolitis
Background of autoimmunity (Rheumatoid Arthritis, Sjogren…)
Small nodules (centrilobular/broncocentric) bronchial wall thickening
Surgical biopsy (BAL is an ancillary test)
Macrolides
Immunodeficiency (familial, common variable immunodeficiency, HIV…)
Steroids
Dyspnea on effort, bronchorrhea
Obstructive impairment
Lymphoid follicles (B cells) around bronchioles
LIP
Background of autoimmunity (Sjogren….)
Centrilobular nodules, septal thickening, ground glass attenuation, cysts
Surgical lung biopsy
Steroids (BAL is an ancillary test; Azathiprine immunohistochemistry/ Cyclophosph.. molecular tests to exclude a monoclonal component)
Dyspnea on effort, cough
Restrictive impairment
Diffuse interalveolar infiltration of lymphocytes (CD3 + cells), lymphoid hyperplasia (follicles consisting of B cells around bronchioles) scattered granulomas
MALT Lymphoma
Mean age 60 yrs
Rounded or segmental shaped consolidations
Surgical biopsy
Chemotherapy
Asymptomatic (minority of cases)
Air bronchogram
CT scan guided or TBB biopsy
Rituximab
B symptoms (fever, asthenia,…) in a minority of cases)
Ground glass opacities Hilar/mediastinal lymphnodes
BAL
Respiratory symptoms (cough, dyspnea
Reticular, perilymphatic opacities
Autoimmune background (Sjogren..) as a predisponent condition
Extrapulmonary involvement In a significant number of cases
Normal PFTs or restrictive Impairment
Search for serum monoclonal component
Lymphocytes with small to medium-sized irregular nuclei, CD19+, (centrocytic-like or monocytoid appearance); plasmocytic differentiation Lymphoepithelial lesions Perilymphatic distribution of the neoplastic infiltrate Light chain restriction
T cell rich B cell Lymphoma (LYG)
Respiratory symptoms (cough, dyspnea, chest pain, acute respiratory failure)
Multiple nodules
Surgical Biopsy
Chemotherapy
Diffuse reticulonodular infiltrates (rare)
Rituximab
Systemic manifestations (fever, malaise, weight loss)
Cavitation (10–25 %)
Extrapulmonary involvement (skin, CNS, kidney…)
Leukopenia or lymphopenia (CD4+ lymphopenia) in about 20–30 % of cases; serologic evidence of prior EBV infection
Perivascular/vascular polymorphous Infiltrate, necrosis of coagulative type
Scattered (or sheets of) large B cells expressing markers of EBV infection; cells relative to the reactive lymphocyte (CD3+, mainly) background is used to grade the lesions
Intravascular B cell Lymphoma
Occurs in older patients
Peripheral wedge shaped lesions; pleural effusion (bilateral); mosaic oligoemia; normal CT aspects/diffuse pulmonary uptake on FDG-PET
Surgical Biopsy
Chemotherapy
Dyspnea; pulmonary hypertension; clinical onset mimicking pulmonary thromboembolism
TBB biopsy
Rituximab
Systemic symptoms (fever…)
Symptoms manifesting an extrapulmonary involvement (CNS, skin…)
Important reduction of PAO2 and PaCO2 inspite of normal lung volumes
A significant increase of LDH; a variant associated with hemophagocytic syndrome has been reported mostly in Asian populations
Intravascular (small vessels, capillaries) neoplastic lymphoid cells (in the majority of cases expressing B markers); the pattern may be misinterpreted as “intersdtitial pneumonitis” or “minimal changes”
Extra-nasal-type NK/T cell Lymphoma
Systemic symptoms (fever, malaise, weight loss)
Nodules or masses (possibly escavated)
Surgical biopsy
Chemotherapy
Respiratory symptoms (dyspnea, cough, acute respiratory failure)
Superimposed infections manifesting with ground glass attenuation or “crazy paving”
TBB biopsy
Extrapulmonary involvement Skin,…)
An opportunistic infection may be the first clinical manifestation
Marked lymphopenia (CD4 + cells); elevated LDH; hemophagocytic syndrome
Angiocentric infiltration of lung tissue by packed lymphoma cells (small, medium sized or angulated or serpentine nuclei) Azurophilic cytoplasmic granules in Giemsa preps
Neoplastic cells are CD2+, CD56+ and cytotoxic molecules (granzyme and perforin) are positive
In situ hybridization for EBV encoded RNA (EBER) is positive in the majority of cases
Reactive pulmonary lymphoproliferative diseases encompass a spectrum of inflammatory and reactive lesions that are often difficult to diagnose since they are difficult to differentiate from other reactive and neoplastic entities. They includes different clinico-pathological patterns: intrapulmonary lymph nodes, nodular lymphoid hyperplasia, follicular bronchitis/bronchiolitis, lymphocytic interstitial pneumonia (LIP) and Castelman’s disease.
Malignant lymphoproliferative diseases are distinguished in Hodgkin’s and non-Hodgkin’s lymphomas (HL and NHL), affecting B or T/NK cells. Malignant lymphoproliferative disorders may arise as primary pulmonary lymphomas (PPL) within the lung parenchyma without evidence of extrapulmonary involvement at diagnosis or in the subsequent 3 months or as secondary pulmonary lymphomas spreading from systemic lymph nodes, through the circulation or from neighbouring sites (e.g. from mediastinal lymph nodes or thymus).
Malignant proliferative diseases occur more frequently in immunocompromised hosts, having in post-transplantated and in HIV ± patients a slightly different clinical and pathological profile from patients with autoimmune disorders or immune competent hosts.
Reactive Pulmonary Lymphoproliferative Disease
Hyperplasia of lymphoid elements, such as intrapulmonary lymph nodes, mucosa-associated lymphoid tissue (MALT) and lymphoreticular aggregates in the terminal bronchioles, may be seen in a variety of lung disease.
Intrapulmonary lymph nodes are distributed at the hilum and occasionally found in the vicinity of the pleura. Hyperplasia of intrapulmonary lymph nodes may be due to a wide spectrum of causes ranging from common hyperplastic and reactive processes to malignant changes. To evaluate the nature of intraparenchimal lymph nodes high-resolution computed tomography (HRCT), positron emission computed tomography (PET-CT) are useful tools, but surgery is necessary to obtain a definitive diagnosis.
Reactive pulmonary lymphoid hyperplasia includes nodular lymphoid hyperplasia and diffuse lymphoid hyperplasia, this last one encompassing the two histological patterns of lymphocytic interstitial pneumonia (LIP), follicular bronchitis/bronchiolitis and Castleman’s disease (CD).
Clinical Vignette
A 37 year-old Nigerian male was admitted to our hospital suffering with cough and fever with chills The patient immigrated from Nigeria 1 years prior to presentation. He reported no exposure to tuberculosis and tuberculosis skin test was negative. In the past he suffered from malaria. Laboratory tests revealed anemia (Hb 6.5 g/dl), elevated C reactive protein (CRP), monoclonal hyperimmunoglobulinemia IgG Kappa, and an elevated interleukin-6 level: 37, (normal value <11). A contrast enhanced CT of the chest and abdomen showed ground glass opacities and interlobular septal thickenings in the lungs (Fig. 31.1) accompanied with axillary, hilar, mediastinal and abdominal lymphadenopathies with hepatosplenomegaly. Rigid broncoschopy was performed for transbronchial needle aspiration in sub carinal lymph node and transbronchial lung biopsy. Plasmacytic infiltration of the lymphoid tissue with scattered lymphoid CD30 positive cells with a blastoid morphoplogy and plasmacytic and lymphocytic infiltration in the pulmonary interalveolar septa and around the bronchovascular bundles were documented. In situ hibrydization using probes to detect the Human Herpesvirus 8 (HHV-8) particles revealed positive cells in the lung parenchyma.
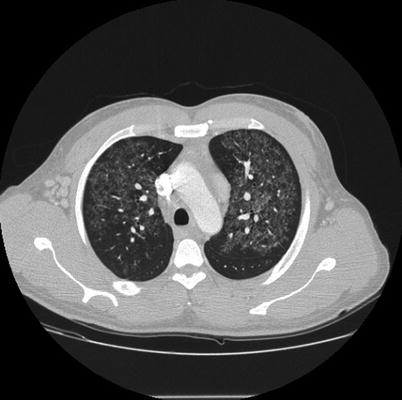
Fig. 31.1
Lung window of the contrast-enhanced CT scan of the chest. Multiple centrilobular ground glass opacities are present in both upper lobes. A relative subpleural sparing and mild septal thickening are also present
To confirm the diagnosis, of Castleman disease an excision biopsy of axillary nodes was performed. Interfollicular expansion composed predominantly of plasma cells with some degree of atypia associated with follicular hyperplasia was documented, suggesting HHV-8 correlated multicentric Castleman’s disease, plasma cell type (Fig. 31.2a, b).
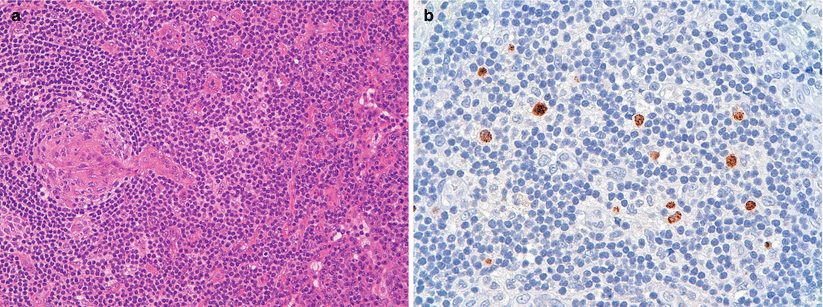
Fig. 31.2
(a, b) Histological finding of axillary nodes biopsy: (a) A burn out follicle with a prominent, hyalinized penetrating blood vessel (lollipop appearance); characteristic “onion skin” appearance due to the lamination of the mantle cell layers. (b) Focal immunoreactivity for HHV8 Interfollicular expansion composed predominantly of plasma cells and some degree of atypia associated with follicular hyperplasia
After chemotherapy, the symptoms that included fever and sweating subsided.
Castleman’s Disease
Castleman’s disease is an uncommon clinicopathological entity first described in 1956 [1]. The incidence is not known and can occur at any age, though it has mostly been reported in adults in the literature. CD is classified according to the clinical and the histopathologic profile, as localized (hyaline- vascular, plasma-cell type) or multicentric with histologic features of one or both of the localized types [2, 3]. The hyaline- vascular type (HV) shows numerous follicles with around a concentric layering of B small lymphocytes (CD20+, IgD+) in an onion-skin apparence, depleted, abnormal, germinal center (CD20+, Bcl6+, Bcl2−) with penetrating hyalinized capillaries in “lollipop” apparence, and large dysplastic cells with vescicular nuclei consistent with follicular dentritic cells (CD21+, CD23+, CD35+). Actually, it has been recognized that CD represent a neoplasm of follicular dentritic cells because have been reported clonal cytogenetic abnormalities in FDC cells [4]. The interfollicular stroma shows marked vascular proliferation, polyclonal plasma cells, immunoblasts, plasmacytoid dentritic cells (CD123+, CD68+,) and myoid cells. Cases of CD in wich the myoid cells are prominent have been referred to as stroma-rich variant of hyaline-vascular CD [5].
The plasma cells type (PC) is characterized by a diffuse polytypic or monotypic (IgA lambda) plasma cells proliferation, often in sheets, in the interfollicular stroma.
The localized form of the disease is mostly asymptomatic with a single site lymph node enlargement. The sites commonly involved are abdomen, peripheral lymph nodes and the mediastinum. It is often discovered incidentally during routine examination, chest X rays. Diagnosis is made by histological analysis of the lymph node biopsy to distinguish it from a thymoma. Multifocal CD, however, presents with systemic symptoms along with multiple lymph node hyperplasia.
Interest in multicentric (M-CD) has grown following the observation that this pathology is often associated with human immunodeficiency virus (HIV) infection and with (HHV8, or Kaposi’s sarcoma herpesvirus KSHV) and Epstein Barr virus (EBV) infections [6]. Pathogenesis remain largely unknown, but HHV8 and Epstein Barr virus may encodes for an homologue of interleukin 6 (vIL 6) which may mediate some systemic features of MCD. Multicentric CD in HIV + patients have a higher prevalence of pulmonary symptoms and usually present with generalized lymphadenopathy characterised by perifollicular vascular proliferation and germinal center angiosclerosis, polyclonal hypergammaglobulinemia, hepatosplenomegaly and constitutional symptoms.
In immunocompetent host the more common pattern is the hyaline-vascular, the associated clinical type is usually the localised one, an asymptomatic mediastinal mass quite benign because in most cases curable by surgery. The plasma-cell variant is more often associated with the multicentric disease characterized by lymphadenopathy, hepatosplenomegaly, skin rashes, sweating, fatigue, anaemia, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia and bone-marrow plasmacytosis. It may be associated with the POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, M-proteins and skin changes). The systemic symptoms are thought to be primarily a consequence of elevated Interleukin-6 (IL-6) production [7, 8]. Recently, a unique clinicopathologic variant of multicentric Castleman’s disease (MCD) called TAFRO (thrombocytopenia, anasarca, fever, reticulin fibrosis and organomegaly) has been identified in Japan. This disease is characterized by a constellation of symptoms, as listed above, and multiple lymphadenopathy of mild degree with a pathologic diagnosis of atypical CD, often posing diagnostic and therapeutic problems for pathologists and hematologists, respectively. These findings suggest that this disease represents a novel clinical entity belonging to systemic inflammatory disorders with a background of immunological abnormality beyond the ordinal spectrum of MCD [9].
Clinical manifestations of lung involvement include dyspnoea, cough, bilateral crackles, or rapidly progressive respiratory failure. Pulmonary symptoms develop always simultaneously with severe systemic manifestations: high grade fever, malaise, generalised lymphadenopathy and hepatosplenomegaly. Hypoxaemia and hypocapnia are often present.
Chest radiographs show reticular and/or nodular interstitial patterns often associated with mediastinal lymphadenopathy and in some cases accompanied by bilateral pleural effusions. Common CT findings are poorly defined centrilobular nodules, thickening of the bronchovascular bundles and interlobular septae, and thin-walled cysts, mostly related to diffuse interstitial, interalveolar, infiltration of small lymphocytes (LIP pattern). Less common findings include subpleural nodules, areas of ground glass attenuation air-space consolidation and bronchiectasis. Bronchoscopy is usually negative, BAL hypercellularity mainly due to an increase of CD8 + lymphocytes is usually present but it is not specific. Microbiological investigations on BAL fluid permit to exclude common infectious aetiologies, and genotyping analysis allows to detect HHV-8 DNA. Definite diagnosis is histological, most cases being diagnosed by peripheral lymph-node biopsy. Specimens of pulmonary lesions may be obtained by trans-bronchial biopsy or by surgical biopsy. Localized CD usually has a good prognosis and requires surgical excision of the enlarged lymph node with no further treatment. M-CD however tends to have a variable prognosis with no documented treatment consensus [2, 3]. Median survival in HIV + patients is 14 months, patients die of infections, transformation to NHL (Large B cell lymphoma associated M-CD or Plasmablastic lymphoma) or chemotherapy toxicity. A variety of combination treatments have been tried with surgical excision, chemotherapy and steroids. In patients with associated Kaposi’s sarcoma polychemotherapy (e.g. cyclophosphamide, vincristine, doxorubicin and prednisone) has been tried with limited success. First line treatment of M-CD in non HIV patients consist of single chemotherapeutic agent, high doses of steroid and/or anti CD-20 monoclonal antibodies. Studies show that humanized anti-human interleukin-6 receptor monoclonal antibody significantly alleviate chronic inflammatory symptoms and wasting [10, 11]. Systemic and pulmonary symptoms improve more rapidly and may disappear. Recently it has been demonstrated that the M-CD symptoms improved by treatment with tocilizumab, which is a humanised antihuman IL-6 receptor monoclonal antibody and corticosteroid [12]. Complete resolution of TAFRO syndrome after immunosuppressive therapies using corticosteroids and cyclosporin A has been reported [13]. However prognosis remain poor, patients die after relapsing of disease, progression to lymphoma or for bacterial sepsis, median survival time being 5 years. Finally treatment with the antiherpesvirus drug, gangciclovir alone or with the antiCD20 B cell monoclonal antibody, rituximab, may markedly improve outcome.
Nodular Lymphoid Hyperplasia
Nodular lymphoid hyperplasia, also known as “pseudolymphoma”, is a localized mass characterized by a lymphoid infiltrate with a lack of evidence of clonality despite immunohistochemical and genetic studies. The most common clinico-radiological feature is a localized and asymptomatic mass, although few patients present fever and elevated ESR. The single lesion is usually curable by surgical excision. Finally it is needed to evaluate the possibility of nodular lymphoid hyperplasia (pseudolymphoma) or clonal lymphoproliferative disorder in the lung as potential manifestations of immunoglobulin(Ig)G4-related disease [14, 15].
Lymphocytic Interstitial Pneumonia (LIP)
Lymphocytic interstitial pneumonia (LIP) [16, 17] is a rare interstitial lung disease characterized by the presence of aggregates of B and T reactive lymphocytes within the lung interstitium.
LIP is associated with serum protein abnormality (monoclonal gammopathy, polyclonal dysproteinemia, hypogammaglobulinemia), immunological disorders, such as Sjogren syndrome (25 % of cases), primary biliary cirrhosis, myasthenia gravis, Hashimoto thyroiditis, pernicious anemia/agammaglobulinemia, autoimmune haemolytic anemia, systemic lupus erythematosus, celiac disease, HIV infection, EBV infection, chronic active hepatitis, other infections (e.g., pneumocystis, tuberculosis), drug injury, allogeneic bone marrow transplantation (GVHD), extrinsic allergic alveolitis. LIP occurs more commonly in women and the mean age is around 55 years.
Presenting symptoms are progressive cough and dyspnoea, weight loss, fever, arthralgias. Common physical findings are bibasilar crackles and finger clubbing (reported in about 50 % of cases). Pulmonary function tests show reduction of lung volume, reduction of DLco, hypoxemia and usually hypocapnia.
The chest radiograph characteristically shows bibasilar reticulonodular infiltrates, a mixed alveolar-interstitial pattern can occur when infiltrates coalesce and cause compression of the alveoli. Typical HRCT abnormalities consist of areas of ground-glass attenuation and poorly defined centrolobular nodules and subpleural small nodules, mostly bilateral (>90 %) and with a diffuse distribution (>60 %). Other common findings are thickening of bronchovascular bundles, interlobular septal thickening (82 %), cystic lesions (68 %) (Fig. 31.3), and lymph node enlargement (68 %). Less common findings include nodules 1–2 cm in diameter (41 %), airspace consolidation (41 %), emphysema (23 %), bronchiectasis (18 %), pleural thickening (18 %), and honeycombing (5 %). Honeycombing and pulmonary hypertension appears in advanced disease. Pleural effusion are infrequent, except in HIV related LIP. Usually the presence of pleurisy, large nodules and mediastinal adenopathy is suggestive for pulmonary lymphoma. Histologically, LIP is characterized by a heavy interstitial lymphoid infiltrate with minor peribronchiolar involvement. Granuloma formation is sometimes noted. Intraalveolar accumulation of small lymphocytes, scanty granulation tissue tufts, and proteinaceous material along with type II cell hyperplasia are ancillary findings. Immunohistochemestry using CD20 shows that B cells are mainly limited to germinal centres (CD10+, Bcl6+, Bcl2−). The interstitial, interalveolar, lymphocytes are prominently T-cells, while the follicles are mainly constituited by B lymphocytes. The immunoglobulin heavy chain gene or the T cell receptor gene using the polymerase chain reaction show no rearrangement. Epstein–Barr virus has been identified in lung biopsy specimens from both HIV infected and non-infected patients [18, 19].
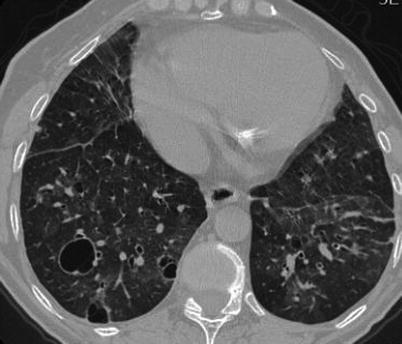
Fig. 31.3
CT scan shows bilateral areas of ground glass attenuation, with a pathcy distribution. Some cyst, variable in size are present in both lower lobes, mainly on the right side. Mild interlobular septal thickening is also present. Findings are consistent with LIP
Treatment with corticosteroid and immunosuppressive drugs may lead to resolution. Median survival is 11,5 years. The outcome is unpredictable and may vary from resolution to death due to progression to fibrosis, cor pulmonale and respiratory failure, to superimposed infection, or to development of a complicating lymphoma.
Follicular Bronchitis/Bronchiolitis
Follicular bronchitis/bronchiolitis is a term introduced to describe the predominant peribronchial lymphocytic infiltrate with abundant germinal centres, often associated with various allergic diathesis, immunodeficiency disorders (HIV infection, common immunodeficiency syndromes), and collagen vascular diseases.
Patients usually present dyspnoea, occasionally fever and cough, hypoxemia, hypocapnia; either obstructive or restrictive spirometric patterns have been reported. The chest film shows bilateral reticular or nodular opacity. Common high-resolution CT findings are centrolobular nodules, bronchiolar dilatation, tree in bud and mosaic perfusion patterns. Expiratory dynamic HRCT scans are important to assess air trapping. Flow-cytometry of BAL usually document a slight increase of polyclonal B lymphocytes. Surgical lung biopsy is often performed to obtain a definite histological diagnosis. Therapy with steroids and also with macrolides at low dose may have some benefit.
Primary Pulmonary Lymphomas
Primary Pulmonary Lymphomas (PPL) are defined as a clonal lymphoid proliferation affecting one or both lungs (parenchyma and/or bronchi) in a patient with no detectable extrapulmonary involvement at diagnosis or during the subsequent 3 months.
The World Health Organization Classification of tumours of lung (WHO 2004) classifies PPL into B-cell primary pulmonary NHL, as Marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT) type, Primary pulmonary diffuse large B-cell lymphoma (DLBCL) and Lymphomatoid granulomatosis (LYG), nevertheless the lung may be the primary site of presentation of most type of nodal lymphoma (WHO Classification 2008) such as Follicular lymphoma (FL), Mantle cell lymphoma (MCL), extraosseous plasmacytoma (EP), Intravascular large B cell lymphoma (IVLBCL), Large B-cell lymphoma arising in HHV8-associated multicentric Castleman disease, Plasmablastic lymphoma (PBL), T/NK lymphoma, Anaplastic large-cell lymphoma (ALCL), and Hodgkin lymphoma (HD).
Primary pulmonary non Hodgkin’s lymphoma (NHL) is very rare and accounts 0.4 % of all lymphomas and 3.6 % of extranodal lymphoma. Mature B-cell neoplasmas are the prevalent fenotype.
B-Cell Non-Hodgkin Lymphomas
The most common histotype is mucosa-associated lymphoid tissue type (MALT) B cell lymphoma, which represents 70–90 % of all primary pulmonary NHL [16–21]. Diffuse large B cell lymphoma (DLBCL) occurs only in 10 % cases of primary pulmonary NHL [21]. In some cases, transformation from MALT lymphoma to DLBCL may occur. Clinically, these present with non-specific symptoms. Radiologically, these can present as consolidation, well-defined mass or nodules. So, PPL can easily be confused radiologically with primary lung carcinoma or metastases when presenting as multiple masses or/and nodules but they have different treatment and prognosis. The main diagnostic criterion for PLL is the absence of extra-pulmonary involvement. Therefore, in patients with biopsy-proven lymphoma of the lung, PLL is diagnosed if extra-pulmonary involvement is ruled out. B-cell primary pulmonary NHL are subdivided into low-grade B-cell PPLs (58–87 %), high-grade B-cell PPLs (11–19 %). The high-grade B-cell PPLs spread rapidly into mediastinal and extra-thoracic locations. This may lead to underestimation of true incidence.
Primary Pulmonary MALT Lymphoma (W.H.O. Classification ICD-O Code 9699/3)
Primary pulmonary MALT lymphoma is a rare extranodal lymphoma that is usually of the low-grade B-cell type and is considered to arise from mucosa-associated lymphoid tissue (MALT) of the bronchus, which is histologically distinct from true intrapulmonary lymph nodes. Low-grade B-cell lymphomas represent 50–90 % of all primary lung lymphomas. MALT-associated malignant lymphomas develop most frequently in the stomach and are also in the bowel, salivary glands, larynx, and thyroid gland [22]. Unlike the model of gastric MALT lymphoma and Heilcobacter pylori, no triggering of antigens has been identified in the primary pulmonary MALT lymphoma. Among the non-gastrointestinal MALT lymphomas the pulmonary lymphomas are the most frequent, (up-to 19 % among MALT lymphomas) [23, 24].
Pulmonary MALT-lymphomas seem to arise on pre-existing inflammatory accumulations of organised lymphoid tissue (lymphoid follicles of the bronchus-associated lymphoid tissue – BALT). BALT is inconspicuous in adults, but the tissue undergoes hyperplasia in patients with chronic immune-mediated diseases such as chronic infections, connective tissue diseases, rheumatoid arthritis, and Sjogren’s syndrome. The cause of these inflammatory processes is likely related to chronic antigen stimulation, as in other extranodal lymphomas, where this correlation (and especially that with infections) is now well established and also relevant for specific therapy [24]. Accordingly, the occurrence of intraclonal sequence variations (ongoing mutations) is a common finding in both gastric and pulmonary lymphomas, indicating the role of antigen stimulations in their pathogenesis [24, 25]. In a proportion of pulmonary lymphomas correlations have been clearly established with conditions where the immune system is abnormally stimulated or deregulated, such as in autoimmune diseases (Sjogren’s syndrome, Hashimoto thyroiditis, systemic lupus erythematodes -SLE, rheumatoid arthritis), or immunodeficiency (primary or acquired). On the other hand, data regarding infections are scanty, and include occasional reports of pulmonary lymphoma with concomitant infectious diseases (Mycobacterium avium complex, Hepatitis–C virus, Helicobacter pylori). Very rarely an association between yellow nail syndrome and MALT lymphoma in the lung has been reported.
About half of the patients with primary pulmonary MALT lymphoma are asymptomatic at presentation, and nearly half of these cases are identified on the basis of abnormal radiological findings by accident. The pulmonary symptoms are non-specific like cough, dyspnea, chest pain, and occasional hemoptysis, but are more common than constitutional symptoms like body weight loss, fever, night sweats, or fatigue. These symptoms may present for several weeks to months before diagnosis [24]. This indolent behaviour can explain why many cases of pulmonary MALT-lymphoma have been previously defined as “pseudolymphoma” [24–27]. Laboratory findings are non-specific and usually normal: only few patients have increased levels of lactate dehydrogenase (LDH) and/or Beta2-microglobulin in the serum and also less frequently a monoclonal band in serum immunoelectrophoresis is found.
Radiologic feature of MALT lymphoma are solitary, well-delineated soft-tissue masses with air bronchogram. Although hilar and mediastinal lymphadenopathy is not a prominent radiologic finding, nodal involvement is documented at pathologic analysis in about the 30 % of cases. HRCT findings include: areas of aveolar consolidation more frequently centred on dilated bronchi, ground glass attenuation, the presence of the “halo sign”, peribronchovascular nodules, “tree in bud pattern”, peribronchovascular thickening and septal lines [28]. The lesions are multiple in more then 70 % of cases. The so called “angiogram sign” previously considered typical of low grade lymphoma in the lung has been observed in other numerous alveolar filling disorders. Radiographic findings may remain unvaried for several years. Cases of endotracheobronchial MALT lymphoma with polypoid features have been reported causing also unilateral lung hypertransparency. MALT lymphomas have generally been reported not to show increased fluorine 18-fluorodeoxyglucose (18-FDG) accumulation on positron emission tomography (PET). The outcome of MALT-type primary pulmonary lymphoma is generally favorable. More than 80 % of the cases have a 5-year survival rate, and the median survival rate has been more than 10 years. The overall survival is better than other types of non-Hodgkin’s lymphoma [16–21]. Clinical features associated with poor prognosis in a series study of primary pulmonary lymphoma included patients over 60 years of age, elevated serum lactate dehydrogenase and elevated serum beta2 microglobuin levels.
Cytogenetic Features and Molecular Pathogenesis
As in other extranodal-MALT lymphomas, an heterogeneous pattern of cytogenetic abnormalities has been demonstrated in pulmonary lymphomas, including aneuploidy (observed in nearly 40 % of cases, with trisomy 3 and 18 being the most common), and specific chromosomal translocations. Translocation t(11;18)(q21;q21) which characterizes about one third of extranodal marginal MALT lymphomas is the most frequent chromosome translocation occurring in pulmonary MALT lymphomas (38.3–41 % in different series). This translocation involves the API2 and MALT1 genes, and can be then directly correlated to the pathogenesis of this lymphoma [29]. Accordingly, API2 is a member of the IAP (inhibitor of apoptosis) gene family, whereas MALT1, a paracaspase of unknown functions, is able to interact with bcl-10 inducing NF-kbeta (nuclear factor K beta) activation. The abnormal fusion of MALT1 with API2 produces chimeric transcripts involved in inhibition of apoptosis, thus contributing to lymphoma development. Interestingly, as previously observed in gastric lymphomas where t(11;18) can serve as a molecular marker for cases not responding to H. pylori eradication, this translocation defines a distinctive clinicopathologic subtype of pulmonary MALT-lymphomas characterised by the absence of any underlying autoimmune disease and lack of plasmacytic differentiation.
Information regarding the occurrence and frequency of other genetic abnormality involved in the pathogenesis of MALT lymphomas such as t(1;14)(p22;q32) are scanty.
Histology Characteristics
At histological analysis the pulmonary structure is effaced by abnormal lymphocyte infiltration, predominantly localised along bronchovascular bundles, interlobular septa and visceral pleura, in a lymphangitic pattern [30]. As MALT lymphomas arising at other sites, pulmonary MALT-lymphoma is formed by the accumulation of clonal lymphoid cells characterised by the morphological and biological features of marginal-zone B-cells. Marginal-zone cells, that are particularly abundant in the spleen, are post-germinal centre lymphocytes with memory functions that migrate from lymphoid tissues to extranodal sites where they can rapidly become antibody producing plasma cells upon stimulation. Morphologically, lymphoma cells are similar to normal marginal-zone cells, exhibiting a spectrum of cytological features (small-round cells, centrocyte-like cells, monocytoid cells), characterised by small and irregular nuclei, inconspicuous nucleoli, and abundant clear cytoplasm. Neoplastic lymphocytes typically accumulate around non-neoplastic lymphoid follicles, forming poorly defined sheets of cells at the periphery of the mantle zones, extending into the lung parenchyma. The presence of reactive follicles, that can be particularly abundant and are presumably pre-existing the lymphoma development, can pose diagnostic problems at morphological and also immunophenotypical analysis. The presence of lympho-epithelial lesions (neoplastic lymphoid cells infiltrating bronchiolar epithelium) is frequent and involve bronchiolar and bronchial epithelial structures. Histologically the differential diagnosis includes all pulmonary diseases characterised by accumulation of lymphoid follicles, and in particular the spectrum of follicular hyperplasia, follicular bronchiolitis, and lymphocytic interstitial pneumonia, as well as, more rarely, hypersensitivity pneumonitis, inflammatory pseudotumor, intraparenchymal thymoma, and Castleman’s disease. For these reasons the use of immunophenotypical and molecular techniques is recommended to substantiate the histological diagnosis of pulmonary MALT lymphoma, especially when the tissue samples are scanty [31].
In a consistent proportion of cases it is possible to demonstrate lymphoplasmacytic differentiation, with a significant plasma cell component exhibiting immunoglobulin light chain restriction. It is possible that at least some cases of primary plasmacytoma of the lung (a rare low-grade tumor of unclear etio-pathogenesis presenting as isolated nodules or diffuse) can in fact be included in the clinico-pathologic spectrum of MALT lymphomas, together with localised pulmonary amyloidosis (another lesion that has been described in association with pulmonary marginal lymphoma).
Clinical Vignette
A 75-year-old man with a history of progressive impairment of general condition, intermittent fever and weight loss was admitted. The peripheral blood picture was unremarkable. Right-sided pneumonia and both-sided pleural effusions revealed by X-ray examinations. Chest CT showed areas of aveolar consolidation with air bronchogram, ground glass attenuation, the presence of the “halo sign” and peribronchovascular nodules (Fig. 31.4a–c). The radiological picture appeared to be definitely deteriorated to a control of chest CT after 40 days and after antibiotic treatment with macrolides. The clinical conditions was deteriorated too. Transbronchial lung biopsies via rigid bronchoscopy were performed but the pathologic examination was inconclusive. Therefore transparietal fine needle aspiration/biopsy samples obtained under CT guide were performed and the pathologic examination noted an histologic pattern compatible with non specific interstitial pneumonia (NSIP). However, this was not harmonized with the clinical and radiological feature (even considering the significant increase of right pleural effusion). Furthermore at this time the laboratory tests revealed anemia (Hb 6.5 gr/dl).
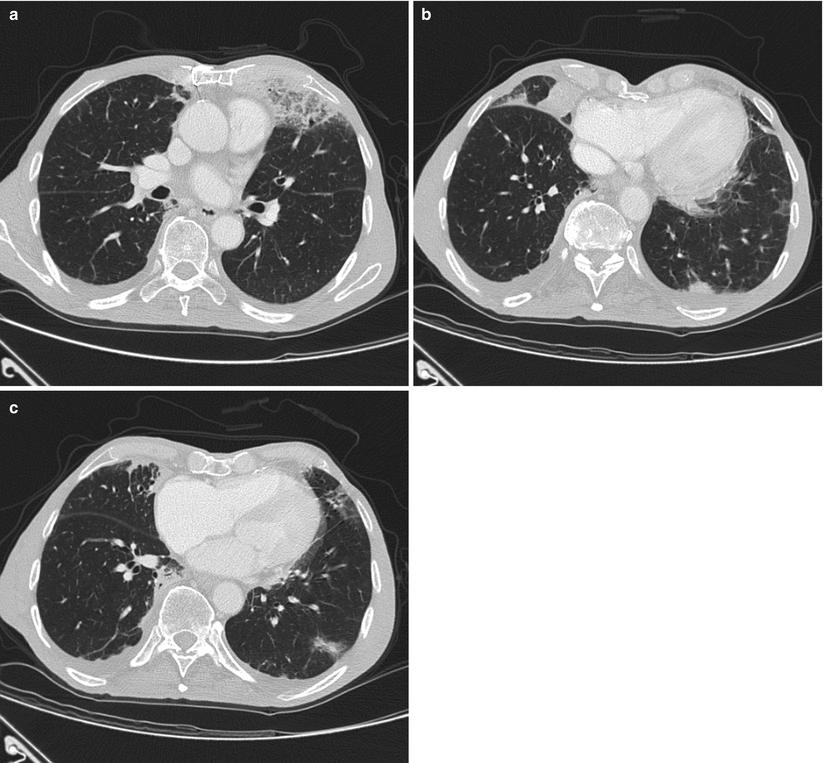
Fig. 31.4
CT scan shows bilateral consolidations, particularly in the middle lobe (b) and in the posterior segment of left lower lobe (b). Moderate ground glass attenuation with interlobular septal thickening is present in the lingula and in the middle lobe. Moderate bilateral pleural effusion
We suspected some lymphoproliferative process and medical thoracoscopic lung biopsy was performed that revealed primary pulmonary MALT lymphoma by immunohistochemical analysis of pleural tissue specimens (Fig. 31.5a–c). Bone marrow examination was negative.
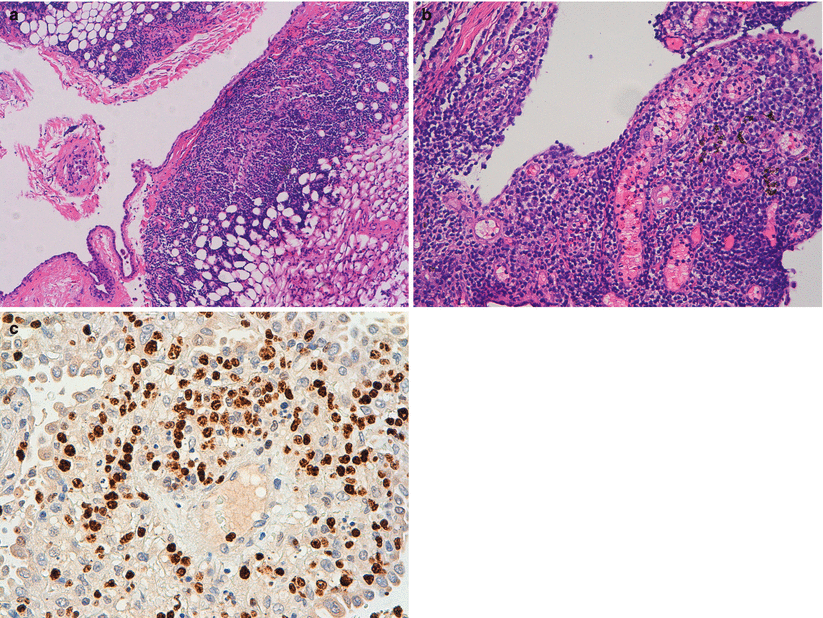
Fig. 31.5
Medical thoracoscopic lung biopsy; (a) In this low magnification view, the neoplastic cells infiltrate extensively the parietal pleura; (b) High magnification view of the pleural neoplastic infiltrate composed of small to medium-sized b lymphocytes with relatively abundant, pale cytoplasm; occasional plasma cells are present. (c) Malt lymphoma with t (11;18) (q21;q21); API2/MALT1 FISH study using a orange probe against MALT1 and green probe against API2. The normal genes appear as isolated orange or green signals, while evidence of the fusion gene (insert) appears as a yellow signal
Pulmonary Plasmacytoma (W.H.O. Classification ICD-O Code 9731/3)
Pulmonary plasmacytoma is a plasma cell malignancy that most commonly occurs in the upper respiratory tract. Plasmacytoma located in the lung is an unusual finding, and in such cases the disease may be confined to the lung and regional lymph nodes or may be disseminated. The most common location for plasmacytoma is the submucosa of the upper airways [32, 33]. It is an extremely rare tumour, less than 50 cases are reported in literature and in fact represent only the 6 % of all extraosseous plasmacytomas. About the 7 % of patients affected by plasma cell myeloma have intrathoracic disease, and it is rarely confined into the lungs. When only located in the lower respiratory tract (primary pulmonary plasmacytoma), diagnosis is difficult and is usually based on the excised tissue
The less differentiated plasmablastic pulmonary plasmacytoma occurs mainly in patients with advanced HIV infection. Phenotypically similar to mature plasma-cells, the malignant cells appear most like plasmablasts. Prognosis in HIV + patients is poor (5.5 months) even if recent small reports suggest it may have improved after HAART advent. The relationship between plasma cell myeloma, solitary plasmacytoma of bone, and extraosseous plasmacytoma is not well understood. For some authors these 3 entities represent different aspects of the same disease spectrum. Others regard solitary plasmacytoma of bone as a rare manifestation of plasma cell myeloma. Extraosseous plasmacytoma should, however, be regarded differently and the diagnosis restricted to tumors that occur outside the bone marrow, may infiltrate nearby lymph nodes or cause distant metastasis. In immunocompetent patients pulmonary plasmacytoma is more frequently observed in the upper respiratory tract. Common clinical findings are cough, dyspnoea and haemoptysis. Laboratory features include paraproteinemia and urinary Bence Jones. When involving lung the most frequent radiologic finding is a pulmonary nodule or mass near the hilum. Lobar consolidation and bilateral diffuse infiltrates have also been described, but this manifestation is very rare [34]. Little is known about endoscopic findings, but as occurred in our third patient, infiltration of the mucosa has been observed, along with polypoid tumors, all lesions which are comparable to those seen in bronchogenic carcinoma [35].
Follicular Lymphoma (W.H.O. Classification ICD-O Code 9690/3)
Follicular lymphoma is generally an indolent B-cell lymphoproliferative disorder of transformed follicular center B cells. Follicular lymphoma is characterized by diffuse lymphoadenopathy, bone marrow involvement, splenomegaly, and less commonly other extranodal sites of involvement such as gastrointestinal tract, lung, skin and other sites [36].
It affects adults (median age 59 years) and it is more frequent in females (male/female ratio 1/1.7). In general, cytopenias can occur but constitutional symptoms of fever, nightsweats, and weight loss are uncommon. Primary lung involvement is usually asymptomatic. HRTC scan shows ground glass opacities sometimes with a “crazy paving” pattern or nodules. Diagnosis is based on histology of preferably biopsy. Immunohistochemical staining is positive in virtually all cases for cell surface CD20, CD10, nuclear for Bcl6 and monoclonal immunoglobulin, as well as membrane expression of bcl-2 protein. The overwhelming majority of cases have the characteristic translocation t(14;18)(q32;q21) involving the IgH/bcl-2 genes. The Follicular Lymphoma International Prognostic Index (FLIPI) prognostic model for FL uses five independent predictors of inferior survival: age >60 years, hemoglobin <12 g/dL, serum LDH > normal, Ann Arbor stage III/IV, number of involved nodal areas >4. The presence of 0–1, 2, and ≥3 adverse factors defines low, intermediate, and high-risk disease with median 10 year survivals in the pre-rituximab era of approximately 71, 51, and 36 months, respectively.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


