Chapter 66
Lymphedema
Evaluation and Decision Making
Stanley G. Rockson
Lymphedema is the clinical description of various disease states characterized by the progressive accumulation of protein-enriched interstitial fluid. These edematous states arise as a consequence of relative impairment of lymphatic drainage. Insufficient lymphatic function can result from either primary or acquired (secondary) anomalies of lymphatic outflow. Cryptogenic forms of lymphedema are often presumed to represent primary lymphatic dysfunction. Although impaired lymphatic function often is manifested as visceral involvement, particularly in the respiratory or gastrointestinal organs, upper or lower extremity edema, with or without visceral involvement, is the most common presentation of lymphatic impairment and is most predictive of the natural history of the condition.
Pathophysiology
Insufficient lymphatic outflow leads to the pathologic end result of lymphedema. In high-input failure, such as that which occurs in venous edema, increased capillary pressure leads to the accentuated production of interstitial fluid; if the production of lymph exceeds the maximal transport capacity of the lymphatic conduits, lymphedema will ensue, even if these structures are anatomically and functionally normal. By contrast, low-output failure ensues when some pathologic condition compromises lymphatic flow. Lymph stasis can accompany lymphatic hypoplasia or aplasia, functional insufficiency or anatomic absence of lymphatic valves, or, conceivably, blunted lymphatic contractility.1 Because the lymphatic circulation provides the normal conduit for the return of interstitial fluid and protein to the central circulation, abnormal lymph stasis creates an accumulation of protein and cellular metabolites in the extracellular space; with the ensuing increase in tissue colloid osmotic pressure, there is water accumulation and elevation of the interstitial hydraulic pressure (see Chapter 13).
Insufficient lymphatic transport leads to the accumulation of hyaluronan and other glycoproteins within the extracellular space. This is followed by a secondary increase in the fibroblast, keratinocyte, and adipocyte content of the affected tissues along with the accumulation of mononuclear cells, including macrophages. Ultimately, an increase in collagen deposition occurs, typically accompanied by an overgrowth of connective tissue and adipose elements in the skin and subcutaneous tissue.2 Although the contributory mechanisms are still not well understood, there is a tendency for these processes to lead to progressive subcutaneous fibrosis.
Classification and Staging
Standard clinical classifications distinguish lymphedema on the basis of cause (primary versus secondary). Primary lymphedema is further classified on the basis of genetics (familial versus sporadic) and time of onset (congenital, praecox, tarda) (Box 66-1).3–5 Although these systems are useful for categorizing lymphedema, they do not address the clinical severity of the disease and are usually not relevant to therapy. More recent classifications focus on the clinical stage of lymphedema or emphasize the underlying anatomic abnormality of the lymphatic system in an attempt to identify the best therapy.1,6,7
Primary Lymphedema
Prevalence of the heritable causes of primary lymphedema is difficult to ascertain, and estimates vary substantially. Primary lymphedema is thought to occur in approximately 1 of every 6000 to 10,000 live births. On the basis of data collected by the Rochester group study, it affects 1.15 per 100,000 persons younger than 20 years.8 Females are affected 2- to 10-fold more commonly than males, and the incidence peaks between the ages of 12 and 16 years.6,9
Of 125 patients with primary lymphedema treated at the Mayo Clinic, 97 (78%) were female and 28 (22%) were male, yielding a female-to-male ratio of 3.5 : 1.10 The ratio of unilateral to bilateral lymphedema was 3 : 1. Congenital lymphedema occurred more frequently in males than in females. In these patients, the edema was usually bilateral and involved the entire lower extremity. In contrast, the typical patient with lymphedema praecox was female and had unilateral involvement, with swelling usually extending up to the knee only.
Primary lymphedema represents a heterogeneous group of disorders; therefore, its classification schemes are numerous. Affected individuals can be classified by age at onset, morphology, or clinical setting.
Classification by Age at Onset and Inheritance
Lymphedema is termed congenital when it is apparent at birth or is recognized within the first year of life. Lymphedema praecox most commonly appears at the onset of puberty, but it may be delayed until the third decade of life. Lymphedema tarda typically begins after the age of 35 years (see Box 66-1).
Congenital.
Congenital lymphedema commonly occurs in a sporadic fashion; however, when clusters of cases occur in families, an autosomal dominant pattern of transmission is frequently observed.11 In addition to the genetic causes of isolated lymphedema, there is a strong association between intrauterine and congenital lymphatic dysfunction and heritable chromosomal abnormalities, including Turner’s syndrome, Klinefelter’s syndrome, and trisomy 21, among many others. In congenital lymphedema, the swelling can involve only a single lower extremity, but edema of multiple limbs, the genitalia, and even the face can be seen. Bilateral leg swelling and involvement of the entire lower extremity are more likely in congenital cases than in other forms of primary lymphedema.10
Lymphedema Praecox.
Lymphedema praecox is the most common form of primary lymphedema, accounting for up to 94% of cases in large series. The name Meige’s disease has historically been reserved for a specific familial form of lymphedema with a recessive pattern of inheritance and typical onset at puberty. Lymphedema praecox displays a marked gender imbalance, with an estimated 10 : 1 female-to-male prevalence.2 The edema is usually unilateral and is limited to the foot and calf in the majority of patients.10 Estrogenic hormones may play a role in the pathogenesis of this form of primary lymphedema.10
Lymphedema Tarda.
Lymphedema tarda is relatively uncommon. Appearing after the age of 35 years, it typically accounts for an estimated 10% of cases of primary lymphedema.
Classification by Morphology
It has been suggested that a morphologic classification of primary lymphedema might provide more useful prognostic information than classification by age at onset (Fig. 66-1).2 This alternative classification scheme relies on an anatomic description of the lymphatic vasculature.3,11
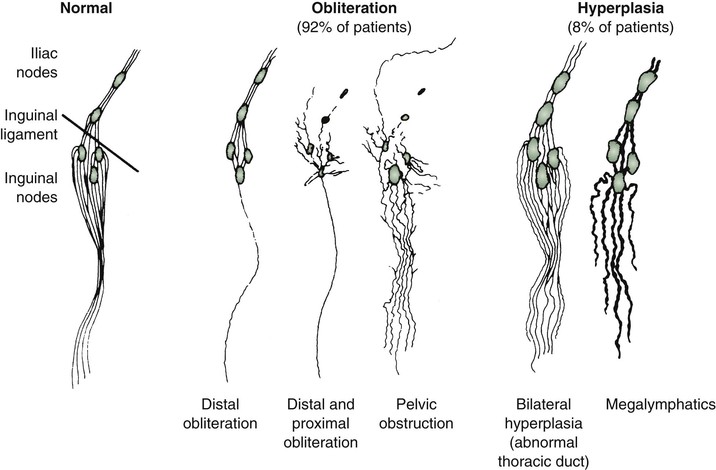
Figure 66-1 Lymphangiographic patterns of lymphatic morphology in a normal lower limb and in patients with different types of primary lymphedema. Obliteration of the lymphatic pathways may be due to aplasia, hypoplasia, or obstruction of the lymphatic channels and nodes.
Aplasia.
In aplasia, no collecting vessels can be identified.
Hypoplasia.
In hypoplasia, a diminished number of vessels are seen.
Numerical Hyperplasia.
In numerical hyperplasia (as defined by Kinmonth3), an increased number of vessels are seen.
Hyperplasia.
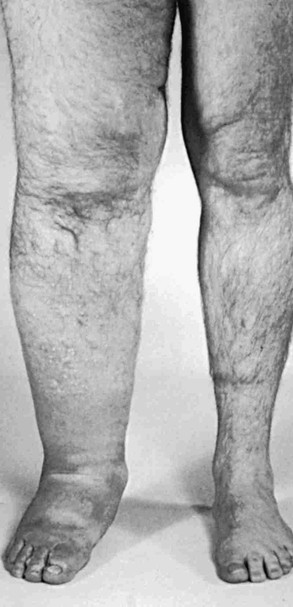
In addition to an increase in number, the vessels have valvular incompetence and display tortuosity and dilatation (megalymphatics, lymphangiectasia). Megalymphatics and lymphatic hyperplasia are less common than hypoplasia or aplasia. This pattern demonstrates a male predominance. These patients most often have unilateral edema involving the entire lower extremity. Cutaneous angiomas and chylous reflux can also be seen (Fig. 66-2). Megalymphatics are associated with a greater extent of involvement and a worse prognosis.
Classification by Anatomy
Aplasia and hypoplasia have a different natural history, depending on whether they involve the distal or proximal portion of the leg.
Distal Obstruction.
Approximately one third of all cases are secondary to agenesis, hypoplasia, or obstruction of the distal lymphatic vessels, with relatively normal proximal vessels (see Fig. 66-1).11 In these cases, the swelling is usually bilateral and mild, and females are affected much more frequently than males. The prognosis is good. In general, after the first year of symptoms, there is little extension in the same limb or to uninvolved extremities. Although the maximal extent of involvement is established early in the disease in about 40% of patients, the girth of the limb continues to increase. Distal hypoplasia or aplasia of the lymphatics most often correlates with the presence of bilateral peripheral edema of the lower extremities. Familial occurrence, female predominance, and indolent progression characterize this pattern of lymphatic disturbance.
Proximal Obstruction.
In more than half the cases, the defect primarily involves obstruction of the proximal lymphatics or nodes, with an initial lack of involvement of distal lymphatic vessels. Pathologic studies reveal intranodal fibrosis.12 In these cases, the swelling tends to be unilateral and severe, and there may be a slight female predominance.11 In patients with proximal involvement, the extent and degree of the abnormality are likely to progress, requiring surgical intervention. Initially uninvolved distal lymphatic vessels may become obliterated over time. A minority of patients have a pattern of bilateral hyperplasia of the lymphatic channels. In these less common forms of primary lymphedema, there is a slight male predominance. When isolated proximal obstructive hypoplasia is observed, clinical involvement of the entire limb is more likely, with relentless worsening of edema.
Classification by Clinical Setting
Alternatively, primary lymphedema can be classified by abnormal phenotype or associated clinical anomalies (see Box 66-1).13
Inheritance.
Although sporadic cases of primary lymphedema are more common,11 the tendency for congenital lymphedema to cluster in families is significant (Fig. 66-3). A familial predisposition for congenital lymphedema, which was ultimately determined to have an autosomal dominant form of inheritance with variable penetrance, was first described by Milroy in 1892.14 He reported “hereditary edema” affecting 22 individuals in 1 family over 6 generations. Although Milroy studied not only congenital lymphedema but also the praecox and tarda variants of the syndrome that bears his name, lymphedema praecox is better known as Meige’s disease.15
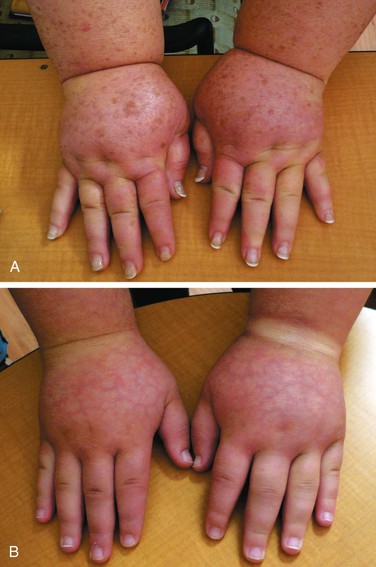
Figure 66-3 A, Adult patient with congenital lymphedema. In addition to the bilateral arm lymphedema depicted, she has edema of both legs and the face. B, Upper extremities of this patient’s 18-year-old son, who has a similar distribution of lymphedema. This is an example of Milroy’s disease.
In general, congenital lymphedema with autosomal or sex-linked recessive forms of inheritance is less common than that with dominant forms of inheritance.11,16,17 Nevertheless, the list of heritable lymphedema-associated syndromes is long and growing.18 Primary lymphedema has been described in association with various forms of chromosomal aneuploidy, such as Turner’s and Klinefelter’s syndromes; with various dysmorphogenic genetic anomalies, such as Noonan’s syndrome and neurofibromatosis; and with a variety of as yet unrelated disorders, such as yellow nail syndrome, intestinal lymphangiectasia, lymphangiomyomatosis, and arteriovenous malformation.19–24 The association of lymphedema with vascular anomalies suggests a common developmental origin of the lymphatic and blood vasculature.
Associated Disorders.
Numerous disorders are associated with heritable forms of lymphedema. Increasingly, these disorders have yielded to chromosomal mapping techniques. Lymphedema-cholestasis, or Aagenaes syndrome, has been mapped to chromosome 15q.25 In several family cohorts of Milroy’s disease, it has been determined that the disorder reflects missense inactivating mutations in the tyrosine kinase domain of vascular endothelial growth factor receptor 3 (VEGFR-3),26,27 thus underscoring the likelihood that this condition reflects an inherited defect in lymphatic vasculogenesis. Several additional lymphedema syndromes have recently lent themselves to successful genetic mapping.7 Lymphedema-distichiasis, an autosomal dominant dysmorphic syndrome in which lymphedema presents in association with a supplementary row of eyelashes arising from the meibomian glands, has been linked to truncating mutations in the forkhead-related transcription factor FOXC228; mutations in FOXC2 have subsequently been associated with a wide variety of primary lymphedema presentations.29 Similarly, a more unusual form of congenital lymphedema, hypotrichosis-lymphedema-telangiectasia, has been ascribed to both recessive and dominant inheritance of mutations in the transcription factor gene SOX18.30 It is plausible that further elucidation of the molecular pathogenesis of these diseases linked to FOXC2 and SOX18 mutations will lead to enhanced insights into the mechanisms of normal and abnormal lymphatic development.
Secondary Lymphedema
Acquired (secondary) lymphedema is the most commonly encountered form of lymphatic dysfunction (Fig. 66-4). In the United States, iatrogenic causes predominate among the acquired forms of lymphedema owing to the common occurrence of lymphatic trauma after surgery or radiotherapy for cancer.9
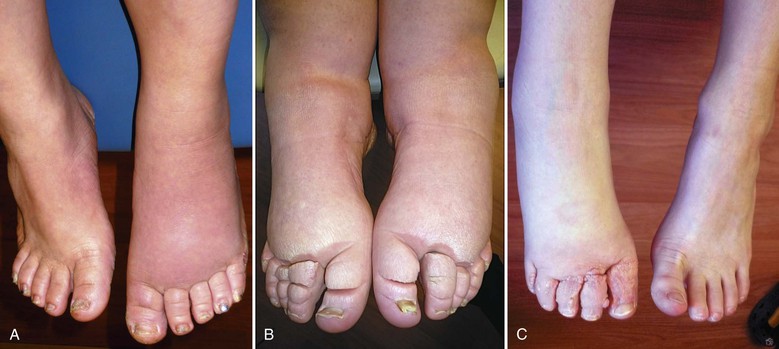
Figure 66-4 Chronic acquired lymphedema of the lower extremities. Note severe skin changes (A) and swelling of the foot (B) associated with squaring of the toes (Stemmer’s sign) and the typical peau d’orange. C, Severe lymphedema with subcutaneous lymph cysts and chronic verrucous superinfection.
Cancer
Of the various clinical settings that predispose patients to lymphedema, treatment of breast cancer is most commonly associated with acquired lymphatic insufficiency (of the upper extremity). Lymph node dissection and adjuvant radiation therapy independently and synergistically predispose to lymphatic vascular insufficiency.20 According to the most recent estimates, 20% to 30% of breast cancer survivors experience clinically significant arm lymphedema after axillary intervention. Despite the benefits of recent surgical and radiotherapeutic enhancements, the problem of lymphedema has not been eradicated.18
Similar lymphatic sequelae are encountered in the lower extremities and pelvis after interventions for gynecologic or urologic malignant neoplasms. Malignant melanoma can cause either upper or lower extremity lymphedema when radical dissection is required in the axilla or groin, respectively.
Filariasis
Filariasis, caused by infestation with parasites such as Wuchereria bancrofti, Brugia malayi, and Brugia timori, is by far the most frequent cause of secondary lymphedema in third-world countries. Of the estimated 90.2 million people in the world who are infected, more than 90% have bancroftian filariasis.31 The disease is most frequent in subtropical and tropical countries such as China, India, and Indonesia. It is transmitted by different types of mosquitoes, and transmission is closely related to poor urban sanitation.32
Perilymphatic inflammation, fibrosis, and sclerosis of the lymph nodes are caused by the indwelling adult worms. Lymph node fibrosis, reactive hyperplasia, and dilatation of the lymphatic collecting channels are caused by the worm products, by physical injury to the valves and vessel walls caused by the live worms, and by the immune response of the host.33 Eosinophilia is found in the peripheral blood smear, and microfilariae can be demonstrated in peripheral nocturnal blood, centrifuged urine sediment, or lymphatic fluid.34 Filarial lymphedema rapidly develops into grossly incapacitating elephantiasis that is extremely difficult to treat.
Other Causes
Lymphedema can also be acquired from other types of lymphatic vascular trauma, including burns and large or circumferential wounds to the extremity. Additional causes of acquired lymphedema include pregnancy, bacterial and fungal infections, infections after snake or insect bites, contact dermatitis, and rheumatoid arthritis.9 Autoimmune destruction of the lymphatics has been hypothesized but not directly demonstrated.
Clinical Staging
Because none of the classification schemes addresses the clinical stage of the disease, the Working Group of the 10th International Congress of Lymphology in 1985 suggested staging chronic lymphedema, regardless of cause. A latent, subclinical stage and three clinical grades were established,35 and each grade was subclassified as mild, moderate, or severe:
Grade III: Edema is irreversible and develops from repeated inflammatory attacks, fibrosis, and sclerosis of the skin and subcutaneous tissue. This is the stage of lymphostatic elephantiasis.
The advantage of this classification is that it permits the evaluation of treatment effectiveness and the comparison of different treatment modalities. One drawback is that appropriate staging may be difficult in some cases without a biopsy of the subcutaneous tissue.
Clinical Presentation
History
A careful history frequently reveals the cause of the swelling and suggests the diagnosis of lymphedema. A family history that is positive for leg swelling may indicate familial lymphedema. The development of painless leg swelling in a teenage girl without any identifiable cause strongly suggests primary (idiopathic) lymphedema. A history of diarrhea and weight loss is a clue to mesenteric lymphangiectasia, whereas intermittent drainage of milky fluid from skin vesicles indicates reflux of chyle. In patients with secondary lymphedema, the cause of limb swelling should be evident from the history, such as previous lymph node dissection, irradiation, tumor, trauma, or infection. In patients who have traveled in tropical countries, filariasis is suspected. Although the causes of primary and secondary lymphedema are different, the clinical presentation and characteristic physical findings are frequently similar.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


