Fig. 17.1
Schematic representation of the mTOR pathway. Tuberin is phosphorylated by multiple inputs from growth signals via growth factors or as a consequence of change in cellular energy status. Phosphorylation of tuberin leads to increased guanine nucleotide hydrolysis via tuberin’s GAP domain. Conversion of guanine triphosphate (GTP) to guanine diphosphate (GDP) inhibits Rheb (Ras homologue enriched in brain) activity; an activator of both mTOR complexes. Activation of the two multiprotein complexes results in differing downstream functions: for TORC1, including the translation of a selection of mRNA species changes in cell size, metabolism, proliferation, autophagy and other functions via the serine/threonine kinase p70S6K and 4EBP1, a component of the protein translation machinery. TORC2 is less well understood but functions include cell migration via the small GTPase Rho
Interestingly, identical genetic abnormalities in TSC-2 have been identified in LAM cells from different sites (lung, lymph nodes, angiomyolipoma) within the same patient; suggesting LAM cells are clonal and migrate throughout the body [10] leading to the ‘benign metastasis model’ of LAM pathogenesis [11]. Consistent with the idea that LAM cells arise from a single precursor which then spread throughout the body, LAM cells have been identified in blood, chyle and urine of patients with LAM [12].
Possibly in keeping with the female preponderance of the disease, LAM cells express receptors for oestrogen and progesterone [13]. In model systems, oestrogen promotes LAM cell growth and metastasis however anti-oestrogen therapies have not proven effective for patients [14, 15]. A major clinical aspect of the disease is the presence of lung cysts. Lined by nodular proliferations of LAM cells, it is thought that cysts may develop as a consequence of extra-cellular matrix proteolysis resulting from the secretion of proteases by LAM cells. Consistent with this idea, it has been shown that LAM cells produce a number of proteases including plasmin [16], cathepsin K [17] and matrix metalloproteinases-1, -2, -9 and -14 [18, 19]. These proteases are capable of degrading extra-cellular matrix proteins including collagens, elastin and proteoglycans. They can also contribute to the disease by activating growth factors, modulating cell surface receptor activity, inflammatory cell trafficking, angiogenesis and cellular invasion. As specific drugs targeting some of these proteins are available they have potential as therapeutic agents. LAM nodules are complex structures comprised of multiple cell types including LAM cells, HMB45 negative stromal cells, lymphatic endothelial cells forming central lymphatic clefts, and covered by hyperplastic type 2 pneumocytes. The interactions between the various cell types and the identity of the stromal cells within the nodules remain to be elucidated. Many aspects of the disease mimic cancer biology and despite the benign appearance of the LAM cell due to their uncontrolled growth, metastatic behaviour, interactions with host cells and a metabolic signature similar to that of cancer cells [20], LAM is viewed by some as a slow growing cancer or cancer-like disease. The processes contributing toward the pathogenesis of LAM are summarised in Fig. 17.2.
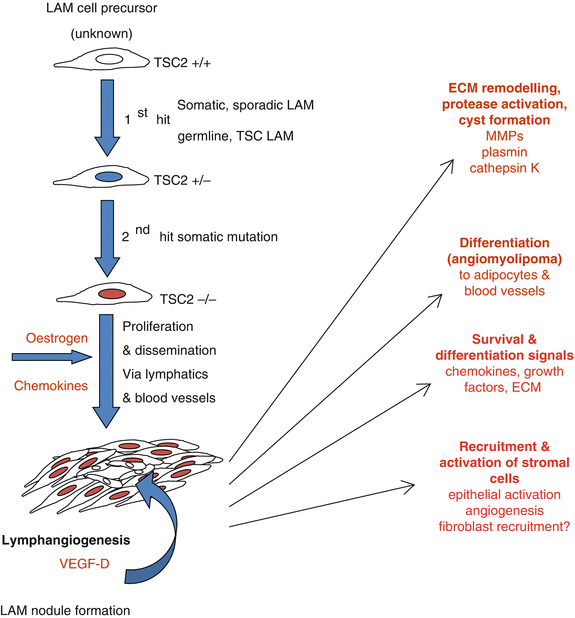
Fig. 17.2
Cellular and pathologic events contributing to the development of LAM. Biallellic inactivation of TSC-2 results in loss of functional tuberin protein in the LAM precursor cell. This confers a survival advantage and metastatic capability which is likely to be oestrogen dependent. LAM cells disseminate and form nodules acting as foci for lymphangiogenesis. The LAM nodule provides a supportive environment for LAM cell growth, is likely to recruit stromal cells, possibly allowing differentiation into the other components of angiomyolipoma including blood vessels and adipocytes. The production of proteases is likely to result in cyst formation and support further LAM cell dissemination. ECM Extracellular Matrix
Presentation
LAM can manifest itself in a number of ways. Most commonly respiratory symptoms are the presenting problem, but abdominal disease, LAM detected as a consequence of TSC and identification in asymptomatic individuals undergoing CT scanning for other problems also occur.
Cysts replace the lung parenchyma to cause breathlessness and symptoms of airway narrowing including cough and wheezing. This collection of symptoms is the presenting feature in over 40 % of patients and frequently leads to treatment for asthma: a poor response to treatment or other features not typical of asthma may then prompt further investigation. Lung cysts also cause pneumothorax which may be recurrent and difficult to treat. Dyspnoea or pneumothorax are the presenting problem in approximately 80 % of patients. Around 5 % of patients present with chylous pleural effusions due to obstruction of the thoracic duct by LAM cells [1]. Chylous effusion and lung cysts in women being pathognomonic of LAM. Some patients cough chylous secretions due to blockage of the intrapulmonary lymphatics whilst others may develop haemoptysis. Onset of respiratory symptoms may occur during pregnancy particularly with refractory pneumothorax including bilateral pneumothorax or chylopneumothorax. Symptoms may persist until surgical correction can be performed, often after delivery [21].
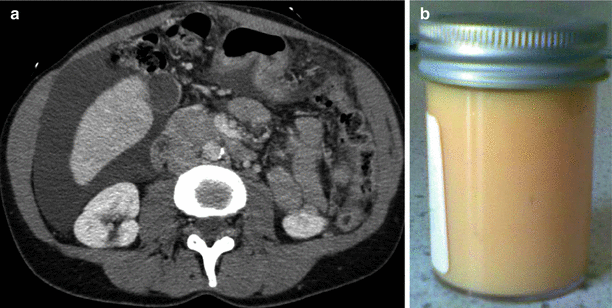
Occasionally abdominal disease may present before lung symptoms. Most commonly this is with symptomatic renal angiomyolipoma. Sometimes large tumours present with abdominal fullness but more commonly haemorrhage causes acute flank pain with or without haematuria. The use of CT scanning to evaluate renal tumours in these situations may coincidentally reveal lung cysts. In some patients, symptomatic angiomyolipoma has preceded lung symptoms by many years. Up to 20 % of patients have cystic lymphatic masses caused by occlusion of abdominal, retroperitoneal or pelvic lymphatics by LAM cells. Termed lymphangioleiomyomas, these can give rise to abdominal bloating, swelling or peripheral oedema [22]. In a small number of cases, discovery of these masses may lead to a biopsy for suspected malignant disease often resulting in chylous leakage with characteristic histology usually leading to the correct diagnosis. In rare cases, symptoms from chylous ascites can be the presenting problem although chylous ascites is generally associated with advanced disease.
Up to 40 % of adult women with TSC have LAM when examined by CT scanning although only a minority of these develop symptomatic respiratory disease [23–25]. The presenting symptoms in TSC-LAM are similar to sporadic LAM with dyspnoea and pneumothorax. Treatment guidelines for TSC recommend screening adult women for TSC at 18 years [26]. This, as well as CT performed for non-respiratory problems in both TSC and sporadic LAM inevitably results in the detection of patients with early and asymptomatic disease. Occasionally, patients with severe learning difficulties may present with advanced disease and even cyanosis or behavioural change due to pneumothorax. The majority of patients with TSC-LAM have renal angiomyolipomas which may be very large, multiple and bilateral and may be the presenting feature [27]. Lymphatic disease appears less common in TSC-LAM than sporadic disease [28]. Clinical presentations suggestive of LAM are listed in Table 17.1.
Table 17.1
Clinical scenarios suggestive of LAM
Asthma with poor response to treatment, especially with fixed airway obstruction |
Early onset emphysema, especially in non-smokers |
Recurrent or bilateral pneumothorax in women |
Pneumothorax in pregnancy |
Chylothorax or chylous ascites |
Respiratory symptoms in TSC |
Angiomyolipoma |
Clinical Vignette
A 41 year old woman developed exertional dyspnoea at the age of 18 and was treated for asthma. Breathlessness worsened over years and was poorly responsive to treatment. Over this period she worked as a technician and gave birth to five children. Some years later she developed abdominal discomfort and became aware of a fullness in her abdomen: irritable bowel syndrome was diagnosed. Four years later, following severe flank pain, she was admitted to hospital. A CT scan showed an 18 cm bleeding angiomyolipoma arising from the right kidney: the tumour was treated successfully by two stage embolisation. The abdominal CT scan also revealed multiple lung cysts in the lung bases and a high resolution chest CT was performed which showed changes consistent with advanced LAM. There were no signs of TSC and TSC gene analysis was normal. Sporadic LAM with renal angiomyolipoma was diagnosed. Lung function showed irreversible airflow obstruction with an FEV1/FVC ratio of 34 % and a gas transfer of 49 % predicted. The history suggests LAM has been present for over 20 years during which time her FEV1 has deteriorated by >100 ml/year. Due to advanced and progressive lung disease she was treated with bronchodilators and Sirolimus.
Diagnosis and Workup
Interstitial changes and preserved lung volumes may be present on chest radiograph (Fig. 17.3) although plain X-rays are often normal at diagnosis. In patients with suspected LAM, high resolution CT scanning is the investigation of choice. The characteristic features are of thin walled cysts. Cysts are evenly distributed throughout the lung fields, are generally round, and vary in diameter between 0.5 and 5 cm. The intervening lung parenchyma is normal although occasionally small areas of airspace shadowing representing haemorrhage or chyle may be present [29] (Fig. 17.4). Widespread alveolar shadowing however is not typical of LAM. Chylous pleural effusions and pneumothorax may also be present (Fig. 17.5). In patients with TSC, nodules of proliferating type 2 pneumocytes, termed multifocal micronodular pneumocyte hyperplasia, may coexist with LAM or occur without LAM [30]. The presence of interstitial abnormalities, thick walled cysts or unevenly distributed cysts is not typical of LAM. CT alone is not diagnostic of LAM and once LAM is suspected, confirmatory features are required to make a definite diagnosis. Either, the presence of renal angiomyolipomas, chylous pleural or abdominal effusions, lymphatics involved by LAM or the presence of TSC. The European Respiratory Society has recently developed clinical guidelines for LAM and their diagnostic criteria are summarised in Table 17.2 [31]. A previous history of renal tumours and symptoms of TSC, should be sought. A careful clinical examination should be made for signs of TSC including the key skin manifestations, facial angiofibromas, periungual fibromas, hypomelanotic macules and shagreen patches. In some patients, these abnormalities are mild and where there is no history of epilepsy or learning difficulties the diagnosis can be difficult to make and evaluation by a TSC specialist or dermatologist may be helpful. Diagnostic criteria for TSC have been clearly defined [32] but where doubt exists referral to a clinical geneticist is advised. To detect the abdomino-pelvic manifestations to aid diagnosis and management, once LAM is suspected contrast CT scanning of the abdomen and pelvis is recommended to detect angiomyolipoma, lymphangioleiomyoma, lymphadenopathy or ascites which are collectively present in over half of patients [31].
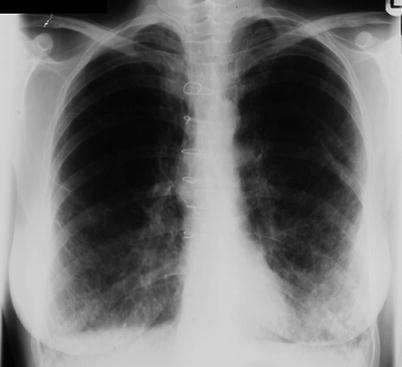
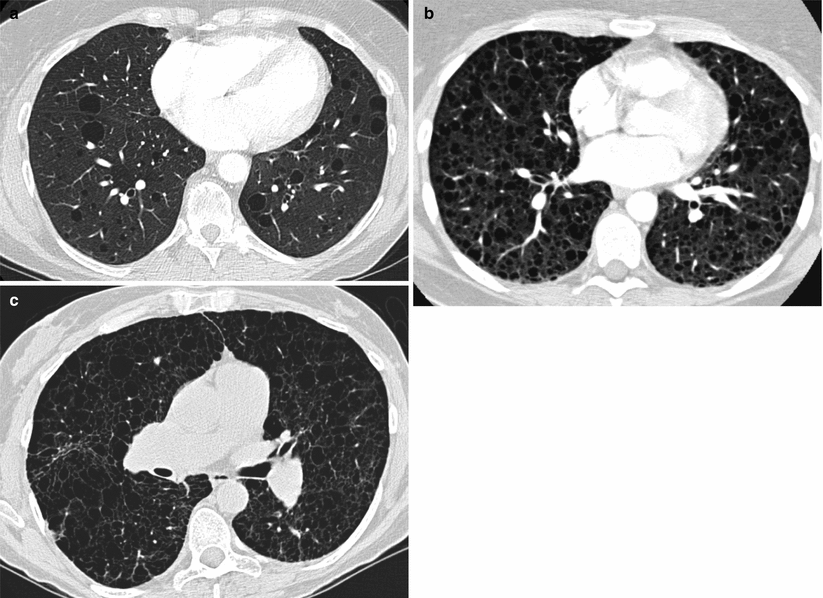
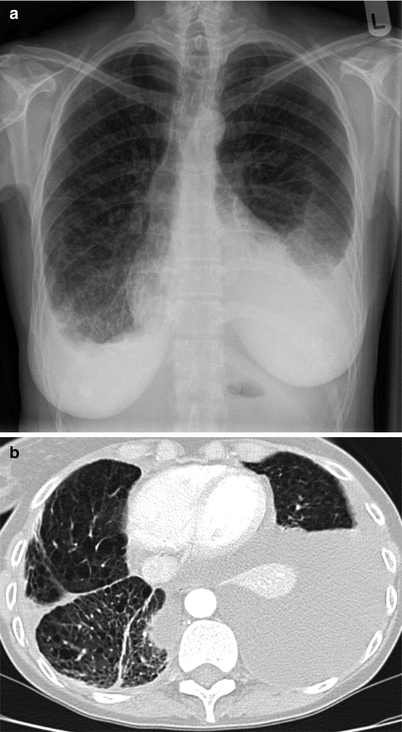
Fig. 17.3
Chest radiograph of a patient with advanced LAM. Reticular shadowing with preserved lung volumes
Fig. 17.4
High resolution CT appearances in LAM. (a) Shows a patient with very slowly progressive disease who has normal spirometry and mildly reduced gas transfer. (b) Shows a patient with progressive LAM with significant airflow obstruction and impaired gas transfer. (c) Shows advanced lung disease with very little lung parenchyma visible
Fig. 17.5
Chylous complications. (a) Chest X-ray and (b) CT from the same patient showing bilateral pleural effusions and parenchymal changes due to LAM
Table 17.2
European Respiratory Society diagnostic criteria for LAM
Definite LAM |
Characteristic or compatiblea lung HRCT, and lung biopsy fitting the pathological criteria for LAMa |
or |
Characteristic lung HRCT and any of the following: angiomyolipoma (kidney); thoracic or abdominal chylous effusion; lymphangioleiomyoma or lymph-node involved by LAM; definite or probable TSCa |
Probable LAM |
Characteristic HRCT and compatible clinical historya |
or |
Compatible HRCT and any of the following: angiomyolipoma (kidney), thoracic or abdominal chylous effusion |
Possible LAM |
Characteristic or compatible HRCT |
Pulmonary function testing may be normal in early disease but a fall in DLCO is generally the first abnormality that develops [33]. As the disease progresses airflow obstruction develops. Lung volumes are generally preserved [34]. Cardiopulmonary exercise testing provides more information on physiological derangement in early disease although is seldom performed [35]. The 6 min walk test probably provides more information about disability and exertional hypoxaemia.
Women with both sporadic and TSC-LAM are at increased risk of meningioma; being present in 8 of 250 patients screened by MRI scanning in one series [36]. Some meningiomas can cause symptoms and require surgery. MRI of the brain may be performed at baseline especially in the presence of headache, seizures or other neurological symptoms. In patients with TSC-LAM and those presenting with LAM who are suspected of having TSC, brain MRI scanning should also be performed [31].
A definite diagnosis of LAM according to ERS criteria can be made without lung biopsy in around 2/3 of patients [37]. In the remainder, a lung biopsy is required to make a firm diagnosis. Whether to perform a lung biopsy or not should be discussed with the patient. In general terms, it is important to obtain a definite diagnosis in patients with progressive disease who require (or may require in the future) specific treatment for LAM. Those with few symptoms and stable lung function may be observed with biopsy being performed if the disease progresses [31]. Lung biopsy may be hazardous for patients with advanced disease and should only be considered if essential to management. Lung tissue may be obtained by transbronchial biopsy and when combined with immunostaining with the monoclonal antibody HMB45 can be diagnostic in some cases and avoid the need for a surgical biopsy [38, 39]. Video assisted thoracoscopic biopsy is performed more often, gives a better indication of the tissue architecture which provides some prognostic information and has better sensitivity and specificity. In most cases the appearance of cysts surrounded by nodular proliferations of mesenchymal cells is sufficient to make the diagnosis in the correct clinical context. In early disease LAM cells may be sparse and their detection can be improved by immunostaining for the smooth muscle markers α-smooth muscle actin and desmin, oestrogen and progesterone receptors [40, 41] (Fig. 17.6). HMB45 stains 30–70 % of LAM cells in biopsy tissue. HMB45 is particularly useful diagnostically, not being expressed in normal lung. Only isolated reports have described cases with the morphological characteristics of LAM in tissues biopsy that do not stain with HMB45.
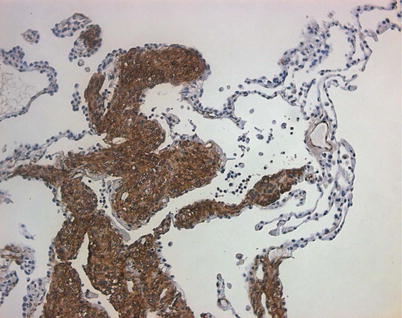
Fig. 17.6
Histological appearance of LAM. Lung section showing lung infiltrated by nodular proliferations of LAM cells which stain strongly for the smooth muscle marker, α-smooth muscle actin (brown)
Recently it has been observed that the lymphatic growth factor, vascular endothelial growth factor-D (VEGF-D) is elevated in around 2/3 of patients with LAM, particularly those with lymphatic involvement [42]. A serum VEGF-D level of greater than 800 pg/ml has been shown to differentiate LAM from other cystic lung diseases and when used in combination with the ERS diagnostic criteria, can further reduce the need for lung biopsy for diagnosis [37, 43]. At the time of writing, the test is not routinely available in all centres.
Prognosis
It is currently difficult to predict prognosis accurately at diagnosis in individual patients. Various studies have associated clinical and pathologic features with outcome and it is likely that presentation with breathlessness rather than pneumothorax, a response to bronchodilators, low KCO at presentation and extensive involvement of the lung biopsy with either cystic change or muscular proliferation are associated with more rapid disease progression [44–47]. In practice, calculation of the disease trajectory by estimating the change in lung function from the onset of symptoms or over a period of observation is probably the most reliable approach. Estimating survival for women with LAM is difficult as older studies have tended to over-represent patients with severe disease and worse outcome and it is important to put these studies into context for patients. Recent studies based on larger patient cohorts have estimated median to be between 20 and 30 years. Improvements in lung transplant outcome and the impact of mTOR inhibitors mean that the prognosis for many patients with a recent diagnosis of LAM should continue to improve.
Management
General Measures
Women with definite or probable LAM are likely to benefit from general measures applicable to other chronic respiratory diseases and should be advised to maintain a normal weight, refrain from smoking, receive prophylactic vaccinations against influenza and pneumococcus and in those limited by dyspnoea undertake pulmonary rehabilitation. Patients with LAM should receive advice on the symptoms of pneumothorax and what to do should these occur. Where relevant, symptoms of bleeding angiomyolipoma should also be discussed. Patients should avoid supplemental oestrogen, particularly in the form of the combined oral contraceptive and post menopausal hormone replacement therapy.
The diagnosis of a rare or orphan disease can lead to a feeling of isolation and helplessness. This may be compounded if incorrect information is given about the disease at diagnosis or the patient is left to find out about the disease themselves. At this time, support from other patients through patient organisations can be very helpful. Strong patient groups exist in many countries including the UK (www.LAMaction.org), France (http://asso.orpha.net/FLAM/), the USA (www.thelamfoundation.org) and others. In addition rare disease organisations such as Orphanet (http://www.orpha.net/consor/cgi-bin/index.php) provide disease specific information for patients.
Parenchymal Lung Disease
Longer term management should be aimed at determining rate of disease progression and avoiding complications. During the course of the disease, lung function, particularly rate of decline of FEV1, DLCO and exercise tolerance should be assessed regularly. Routine follow up, including spirometry and gas transfer is generally scheduled between one to four times a year, with the interval between follow up dependent upon the individual patient’s previous rate of disease progression. On average patients lose FEV1 by around 120–150 ml/year [34, 48], those with loss of lung function due to progressive parenchymal disease, rather than pleural complications, should be seen more frequently and therapy with an mTOR inhibitor considered.
Pleural Disease
Patients with LAM are at high risk of pneumothorax. Pneumothorax occurs in 70 % of patients and is recurrent in the majority of these. On average, patients have four pneumothoraces with each episode requiring 7 days in hospital [49]. Surgical intervention reduces recurrence rates and should be considered after the patient’s first pneumothorax. Evidence is only available from case series but suggests that surgical approaches may be more effective than pleurodesis via chest tube [21, 49]. In a significant number of cases, more than one surgical procedure may be required. There is no clear evidence to suggest one procedure is superior to another in patients with LAM. It is therefore appropriate to perform the minimal degree of pleural intervention which will prevent recurrence. Although pleural surgery results in increased peri-operative bleeding during transplant procedures, it does not seem to affect overall survival [50] and patients with pneumothorax should be treated with the most appropriate surgical procedure to treat pneumothorax.
Clinically significant chylous pleural effusions affect around one in ten patients. Occasionally these are stable and can merely be observed. Simple drainage usually results in rapid reaccumulation of the fluid [51]. Rates of fluid formation may be reduced by a low fat, or medium chain triglyceride, diet however in many cases surgical pleurectomy and possibly thoracic duct ligation may be required. Recently, use of the mTOR inhibitor rapamycin has been shown to reduce the volume of chylous pleural effusions and reduce the need for thoracocentesis in these patients [52].
Renal Angiomyolipoma
Patients with angiomyolipomas should have their renal tumours monitored regularly. Once initial cross sectional imaging using either CT or MRI has been performed, follow up imaging in uncomplicated cases, where a straight forward measurement of growth is required, may be performed by ultrasound (Fig. 17.7). For small tumours with a low risk of bleeding, renal imaging once a year is recommended. For tumours at higher risk of bleeding: specifically, those greater than 4–5 cm in their longest axis, those with aneurysmal blood vessels and symptomatic tumours should be imaged at 6 monthly intervals [53]. Large, enlarging and symptomatic tumours should be evaluated by a urologist, ideally with expertise in conservative management of these lesions. As angiomyolipomas are frequently bilateral, selective treatment of large and symptomatic lesions may be performed by conservative nephron sparing surgery or by selective transcatheter embolisation rather than nephrectomy. Outcomes are similar between techniques although embolisation may be performed without the use of a general anaesthetic including during episodes of haemorrhage and pregnancy [54]. Those with TSC-LAM almost always have renal angiomyolipomas. These tend to be bigger and more likely to bleed than those in patients with sporadic LAM [27] (Fig. 17.8).
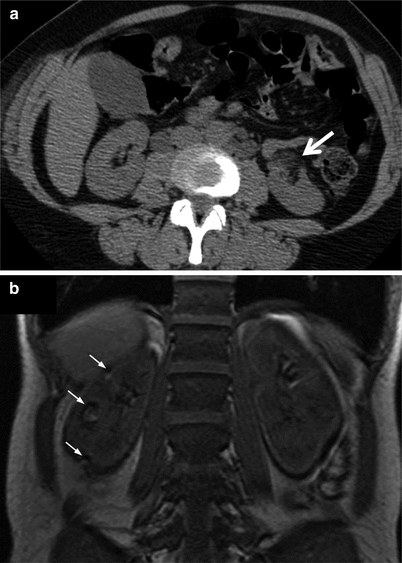
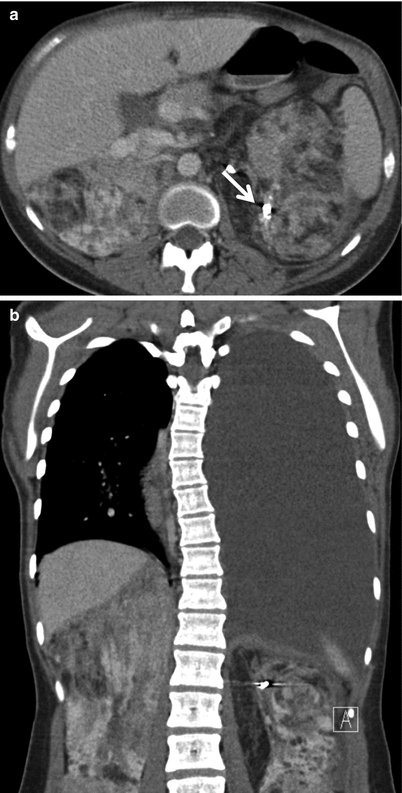
Fig. 17.7
CT appearances of angiomyolipoma in patients with sporadic LAM. (a) Shows a characteristic small asymptomatic lesion in the anterolateral aspect of the left kidney (arrow). The low density areas containing fat are characteristic of angiomyolipoma. (b) Coronal section of a T1 weighted MRI image showing multiple small angiomyolipomas in the right kidney (arrows)
Fig. 17.8
Angiomyolipomas in TSC-LAM. (a) Shows a cross sectional image of a patient with TSC-LAM and multiple, bilateral angiomyolipomas greatly enlarging both kidneys. The arrow highlights an embolisation coil, used to treat a bleeding lesion. (b) Shows a coronal CT of the same patient who presented with dyspnoea due to a large left chylous effusion
Abdominopelvic Lymphatic Disease
Occlusion of the axial lymphatics by LAM cells can result in enlarging cystic structures. Although often asymptomatic, these lesions can be associated with abdominal distension and bloating [55]. Characteristically these lesions enlarge throughout the day, which can be associated with worsening symptoms in the afternoon [56]. Rarely, larger lesions can cause pressure symptoms on other organs including the bladder. Abdominal lymphatic disease may be associated with chylous ascites which can also cause abdominal symptoms (Fig. 17.9). Surgical treatment of abdominal lymphatic masses can be followed by prolonged chylous leakage and is best avoided. Symptomatic measures include reducing fat intake as for chylous pleural effusions. Recently case reports and a case series have suggested that treatment with mTOR inhibitors is effective for symptomatic abdominopelvic lymphatic disease resulting in resolution of symptomatic chylous ascites and lymphangioleiomyomas [52].