2

Lung Growth and Development
This chapter describes the morphologic features and molecular regulation of mammalian and, more specifically, human lung development. In the short time interval between the previous and current editions of this book, molecular biology and genetic experimental approaches have resulted in a vast expansion of our understanding of the molecular regulation of lung development. As predicted by the late William M. Thurlbeck in a previous edition of this book, a significant portion of this chapter consequently is devoted to recent advances in our knowledge of the molecular regulation of lung development, both with respect to early branching morphogenesis and subsequent distal lung remodeling.
Much information about the complex and highly coordinated processes that control lung development is based on genetic studies in Drosophila melanogaster and mouse models, and experimental manipulation in larger animal models such as the fetal lamb and prematurely delivered monkeys. Although these experimental models have significantly advanced our understanding of the molecular and cellular basis of lung development, the clinical relevance for human developmental anomalies and perinatal lung diseases remains to be fully elucidated. It is expected that future editions of this chapter will contain considerably more information regarding the molecular and genetic pathogenesis of human congenital and acquired lung diseases.
This chapter further summarizes the major hormonal, humoral, and physical factors that affect postglandular lung remodeling and alveolarization. Additionally, I have highlighted various conditions associated with pulmonary hypoplasia. The failure of normal distal lung development characteristic in pulmonary hypoplasia is a frequent finding in perinatal and neonatal autopsies. I have included a section describing the practical, systematic approach to examination of the perinatal lung, including standard techniques (determination of morphologic developmental stage, lung/body weight ratio, and radial alveolar count), as well as more advanced and specialized techniques.
Structural/Morphologic Aspects of Lung Development
The respiratory system consists of branched tubes that conduct air to a vast terminal gas-exchange system composed of alveoli. From a functional-molecular point of view, three distinct stages of lung development are identified: initial bud formation; dichotomous branching, which produces the air-conducting system; and formation and maturation of the terminal elements to produce the gas-exchange surface. Lung development so defined conforms precisely to the classical stages of organogenesis namely induction, morphogenesis, and differentiation.1
Using morphologic criteria, lung development can be classified into five different stages.2 The induction phase is called the embryonic stage and is characterized by the formation of the lung bud. This epithelial diverticulum protrudes ventrally from the foregut into the surrounding mesenchyme and branches into left and right primary bronchial buds. This sequence is followed by the pseudoglandular stage, during which the bronchial buds undergo rapid dichotomous branching via complex epithelial-mesenchymal interactions. The pseudoglandular stage is characterized by the formation of the conductive airway tree down to the terminal bronchioles. This sequence is followed by the canalicular stage, when further expansion of the airway tree and massive organ growth occurs along with differentiation of the airspace epithelium. During this stage, the number of capillaries in the primitive mesenchyme increases dramatically. The main gas-exchange surface is formed during the final stages of differentiation: the saccular and alveolar stages.
Stage | Postconceptional Age | Major Events |
|---|---|---|
Embryonal | 21 days to 8 weeks | Organogenesis; development of trachea, major, and segmental bronchi |
Pseudoglandular | 8 to 16 weeks | Development of remaining bronchial tree up to level of terminal bronchioles; differentiation of respiratory epithelial cells; angiogenic development of preacinar vasculature |
Canalicular | 16 to 26–28 weeks | Development of vascular bed and framework of acinus; flattening of epithelium; differentiation of type I and type II alveolar epithelial cells; development of distal pulmonary circulation by vasculogenesis; formation of air–blood barrier; appearance of surfactant |
Saccular | 26–28 to 32–36 weeks | Increased complexity of terminal saccules by secondary crests; decrease in interstitial space; double capillary layer |
Alveolar | ≥ 32–36 weeks to 2 years of age | Alveolarization by septation; microvascular maturation; fusion of capillaries |
The cranial segments of the lung mature faster than the caudal segments, contributing to partial overlap among different authors in the gestational age between the various postglandular stages. Although the division of lung development into five histologic stages is somewhat arbitrary and may appear artificial, as lung organogenesis is a continuous process, it emphasizes the concepts of transitional or progressive phases of fetal lung development.
Morphologic Stages of Lung Development (Table 2–1)
Embryonal Stage (21 Days to 8 Weeks Postconception)
The human lung originates as a ventral diverticulum (laryngotracheal groove) from the wall of the primitive foregut in week 3 of embryologic development, when the embryo is 3 mm long. The laryngotracheal groove separates dorsoventrally from the primitive esophagus to form the tubular lung bud; the upper portion develops into the larynx and the lower portion into the tracheobronchial tree. At the same time, the lung bud branches to form the left and right primary bronchial buds, which eventually develop into the left and right bronchial trees (Fig. 2–1). By the fifth week, elongation, branching, and budding of the two bronchial buds give rise to three bronchial stems on the right and two on the left—the basis for the lobar organization of the mature lung. The embryonal stage ends with the development of these presumptive bronchopulmonary segments.
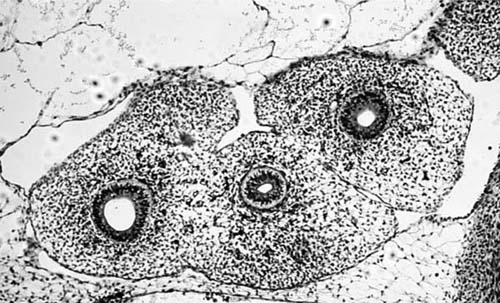
FIGURE 2–1 Embryonal stage (7 weeks). Trachea with left and right bronchial buds surrounded by splanchnic mesenchyme. (Courtesy of Dr. C. E. Oyer.)
The bronchial buds are lined by a thick endodermally derived epithelium, which differentiates into specialized cells that line the conducting and respiratory airways. Mesenchymal cells surrounding the primitive airways give rise to blood vessels, smooth muscle, cartilage, and other connective tissues of the lung. At the end of the embryonic period, the primitive pulmonary arteries begin to form from the sixth aortic arch.3,4 The pulmonary veins form as evaginations from the left atrium during week 4 of gestation and fuse with the mesenchymal capillary plexus in week 5.5,6
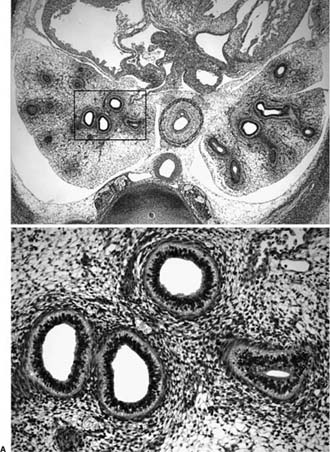
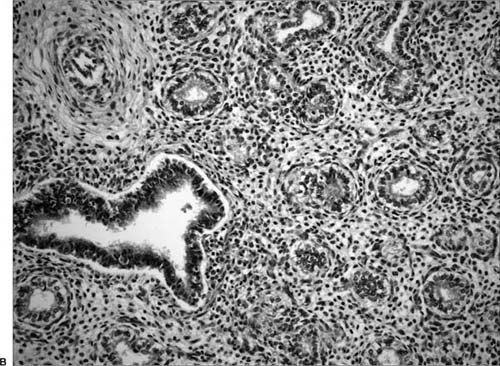
FIGURE 2–2 Pseudoglandular stage. (A) Early pseudoglandular stage (10 weeks). Simple tubular structures branch out within a primitive mesenchymal matrix. Area indicated by rectangle in upper figure is shown at higher magnification in lower figure. (B) Late pseudoglandular stage (16 weeks). Branching pattern is completed, airway lumina are lined by columnar epithelial cells. Ciliated cells are present in the larger airways.
Pseudoglandular Stage (8 to 16 Weeks)
During this stage of branching morphogenesis, further centrifugal branching of the primitive airways into the thoracic mesenchyme generates the presumptive conducting portion of the respiratory system to the level of the terminal bronchioles7,8 (Fig. 2–2). By 16 weeks, all airway divisions are more or less complete, resulting in 12 to 17 branches in the upper lobes, 18 to 23 branches in the middle lobe, and 14 to 23 branches in the lower lobes.7 At the end of this stage, the branching pattern is essentially that of the adult lung.8
The term pseudoglandular is derived from the histologic appearance of the lungs, which on cross section consist of hollow gland-like structures surrounded by a dense cuff of mesenchymal cells. Differentiation of the mesenchyme and epithelia begins in the more proximal regions of the airways and progresses distally. Initially the airway lumina are very narrow, with a thick pseudostratified epithelial lining. From week 13 onward, the lumina enlarge and the epithelium thins to more columnar cells. The pluripotent epithelial cells focally differentiate to ciliated cells and goblet cells. During this stage, cartilage begins to form around the larger airways, and smooth muscle forms around airways and major blood vessels.8
Lymphatics first appear in the hilar region of the lung in gestational week 8 and in the lung itself by week 10.9 The pulmonary arteries develop in parallel with the conducting portion of the lungs and follow the same branching pattern. These vessels are originally connected to systemic veins that drain the trachea and foregut, but they subsequently anastomose with the pulmonary veins arising from the cardiac atrium. At the end of the pseudoglandular stage, all major elements of the conducting system are formed, although gas exchange is not yet possible.
Canalicular Stage (16 to 26/28 Weeks)
In the canalicular phase, acinar development begins with the formation of primitive airspaces and the apposition of capillaries to the wall of these structures, resulting in the formation of a primitive potential air–blood barrier (Fig. 2–3). The terminal bronchioles divide to form two respiratory bronchioles, which in turn branch to form three to six primitive alveolar ducts ending in terminal sacs. This stage is also marked by extensive angiogenesis within the peripheral respiratory mesenchyme, forming a dense capillary network and leading to vascularization of the developing respiratory structures. The potential airspaces are thus “canalized” and approximated by a network of capillaries.10
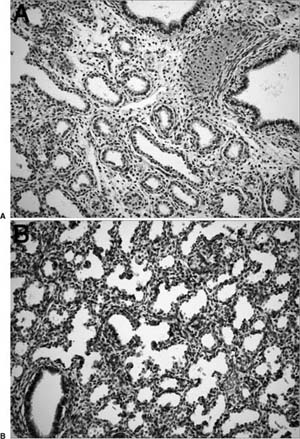
FIGURE 2–3 Canalicular stage. (A) Early canalicular stage (18 weeks). Simple smooth-walled airspaces are separated by abundant, sparsely vascularized, interstitial tissue. (B) Later canalicular stage (24 weeks). The inner lining of the airspaces has assumed a wavy configuration. The epithelium is flatter than in Fig. 2–3A.
Thinning of the mesenchyme continues through this period, and the epithelial lining in the most peripheral parts of the lung begins to attenuate. Flattening of the acinar epithelium at about 20 to 22 weeks marks the initial differentiation of type II pneumocytes. The type II cells retain the cuboidal shape of their precursors and contain lamellar bodies and glycogen in their cytoplasm. The type I cells, which differentiate from type II cells, begin a squamous-like flattening and attenuation to facilitate a potential air–blood interface. The cuboidal intermediate cells of the lower airways differentiate to form ciliated cells and nonciliated Clara cells.
By the end of the canalicular stage, a potentially functional if rudimentary blood–gas barrier has formed, sufficiently thin to support gas exchange. The conducting airways have developed both goblet and ciliated cells and the epithelial cells produce fetal lung liquid. The blood vessels develop along conducting airways and are muscularized more distally than in the adult, and lymphatic structures now begin to appear. Because cranial segments of the lung mature faster than caudal segments, this stage partially overlaps with the next one, extending from 16 to 26–28 weeks’ gestation.
Terminal Sac (Saccular) Stage (26/28 to 32/36 Weeks)
At the end of the canalicular stage, airways terminate in large smooth-walled cylindrical structures. At the onset of the terminal sac stage, vascularized ridges called secondary crests protrude into the terminal saccules, pulling a capillary network with them and causing branching of the saccules, which eventually differentiate into alveolar complexes (Fig. 2–4). The secondary crests contain a double-walled capillary system. The division of the saccules into smaller units by the secondary crests associated with a marked decrease in interstitial tissue and a further increase in the capillary bed results in the development of a complex capillary network in the saccular wall. This ensures efficient gas exchange as alveoli begin to develop at the end of this stage. The major changes occurring during the saccular stage thus include the decrease in thickness of the interstitium, the thinning of the epithelium, and the beginning of septation of the terminal air units. The term saccular stage derives from the sac-like appearance of the most peripheral airspaces.
As vascularized septa form within growing terminal sacs, type I cells continue to flatten and spread, increasing the surface area available for gas exchange. The parenchyma of the lung continues to thin out, and fibroblasts lay down the collagen and elastin fiber components of the interstitium. During this period, type I and type II alveolar cells continue to differentiate, and these alveolar epithelial cells become the most abundant epithelial cells in the lung. Type II cells expand in size and number, accumulate surfactant lipids, and decrease cytoplasmic glycogen. By week 36, the stroma of the lung has thinned to the extent that capillaries may protrude into the prospective alveolar airspaces. The end result of the saccular stage is a rapid increase in the gas-exchange surface of the lung and rapid thinning of the interstitium.
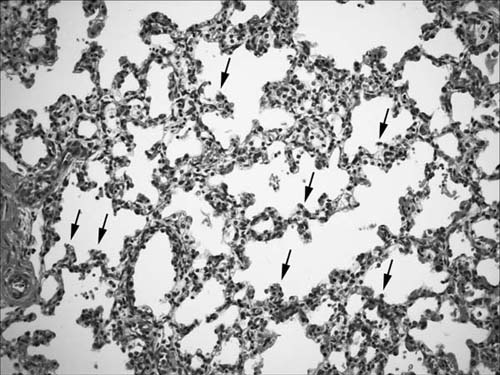
FIGURE 2–4 Terminal sac (saccular) stage (34 weeks). Airspaces are being subdivided by secondary crests, indicated by arrows.
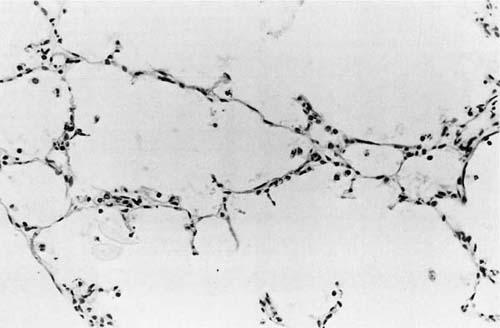
FIGURE 2–5 Alveolar stage (36 weeks). Thin-walled alveoli are readily visible. (From Langston C, Kida K, Reed M, Thurlbeck WM. Human lung growth in late gestation and in the neonate. Am Rev Respir Dis 1984;129:607–613. Used with permission.)
Alveolar Stage (32/36 Weeks to Infancy)
The final aspects of maturation of the respiratory system take place during the alveolar stage (Fig. 2–5). Alveoli develop as flask-shaped structures with thin walls whose double capillary network fuses to appear as a single capillary bed. Mature alveoli are distinguished from terminal airspaces formed by secondary crests because alveoli have thin-walled interalveolar septa with a single layered capillary network and are polygonal, rather than round. Alveolar formation is closely linked to the deposition of elastin in the saccular lung.11
There has been much debate about when the alveolar stage begins, in part because of conflicting definitions of what constitutes an alveolus. Studies by Langston et al11 have shown that alveolar structures can be recognized in some fetuses at 32 weeks of gestation and are present uniformly by 36 weeks, challenging previous beliefs that alveoli develop only after birth.12 Based on this study,11 the general consensus currently is that the alveolar stage commences in utero during the 36th week of gestation. Similarly, there has been controversy about the end of the alveolar stage. Although alveolar development starts late in gestation, the current concept is that the majority of alveoli form during the first 2 years of life. By 3 years, the overall morphology of the lung is well established. Subsequent increase in lung volume occurs through a proportional growth of all distal lung components until adulthood.
By the end of the alveolar stage, lymphatic channels are distributed around pulmonary arteries, bronchi, and bronchioles, and extend along interlobular septa to anastomose with a plexus beneath the pleura.9 The vascular supply of the lung undergoes profound alterations during development. In older children and adults the bronchial arterial circulation originating from the aortic arch supplies the bronchi, bronchioles, and interlobular septa. This is in contrast with fetal life and early infancy, when the bronchial artery also contributes substantially to the circulation of the alveolar ducts and alveoli in the central lung parenchyma through bronchopulmonary artery anastomoses.3
Postnatal Lung Growth
As reviewed earlier in the chapter, alveoli begin to form during late gestation so that alveolarization is well under way at the time of birth. At birth, the number of alveoli is estimated to range from 10 million to 150 million (average: 50 million),11,13 providing a gas-exchanging surface of about 3 to 4 m2. The number of alveoli increases rapidly after birth, reaching the adult range of 300 million to 600 million (surface area 75 to 100 m2) by 1.5 to 2 years of life.14,15 As in intrauterine life, alveolarization and increases in the internal surface area of the lung occur through the protrusion of numerous short, blunt tissue crests or ridges into alveolar sacs. In agreement with Thurlbeck’s16 concepts, secondary septa are rarely seen by 1.5 years in human infants,14,15 suggesting that alveolarization is mostly complete by this time. After 2 to 5 years, the lung continues to grow in proportion to body growth. This later lung growth is mainly an increase in alveolar size and volume rather than further increase in alveolar number.
Septal maturation continues beyond the alveolar stage during what is sometimes called the (sixth) stage of microvascular maturation. This phase extends until 2 to 5 years of age14 and is associated with rapid growth of the arterial bed.17
Differentiation of Airway Epithelium
Maturation of the epithelium starts in the proximal airways and progresses distally into the intrapulmonary airways.18 The proximal-distal arrangement of the various epithelial cell lineages becomes morphologically apparent during the pseudoglandular stage.19 The conducting and respiratory portions of the lung contain at least 11 different epithelial cell types.18 The pseudostratified portion of the proximal tracheobronchial epithelium is mainly composed of basal, secretory and ciliated cells. Type I and type II alveolar cells make up the distal respiratory epithelium. The lineage relationships between the various epithelial cell types have not been fully delineated. Evidence from cell kinetic studies suggests that basal cells, Clara cells, and type II alveolar cells may be the primary progenitor cells for the pulmonary epithelium.20–25 Although pulmonary neuroendocrine cells differentiate morphologically before any other epithelial cell type,26,27 the lineage relationship between neuroendocrine cells and the putative stem cells (type II cells and Clara cells) has not been elucidated. The approximate timing of differentiation of human airway epithelium is shown in Table 2–2.
The onset of cellular differentiation is characterized by the expression of differentiated gene products. Early on in embryonic development, the undifferentiated epithelium coexpresses several lineage markers, including surfactant protein A (SP-A, a marker of alveolar type II cells), Clara cell secretory protein (CCSP, a marker of the proximal Clara cells of the bronchial epithelium), and calcitonin gene–related peptide (a marker of neuroendocrine cells).28 During the pseudoglandular stage, pulmonary epithelial cell linages become restricted to proximal and distal regions of the airways.
Gestational Age (Weeks) | Cellular Events |
|---|---|
4 | Primitive epithelial cells |
8 | Neurosecretory cells |
10–12 | Presecretory and preciliated cells |
12 | Mucous glands |
14 | Neuroepithelial bodies, ciliated cells, and prebasal cells |
16 | Goblet, serous, and basal cells Pre–type II cells and pre–Clara cells |
24 | Type I and type II alveolar cells |
24–26 | Clara cells |
From Cutz E. Cytomorphology and Differentiation of Airway Epithelium in Developing Human Lung. Edinburgh: Churchill-Livingston, 1987. Used with permission.
Comparative Anatomy
The chronologic sequence of staged lung development is highly preserved among mammalian species. In all mammals studied, pulmonary alveoli are formed, in part, by septation of alveolar saccules.11,14,15,17,29–36 However, the time point in development at which septation occurs varies considerably among species in a manner consistent with the activity level of the newborn (Fig. 2–6). For example, guinea pigs33 and range mammals,29,32,34 which have great locomotive capacity at birth, septate in utero (precocial development); others, such as rats35,36 and mice,30 that have little locomotive capacity at birth septate postnatally (altricial development). As noted earlier, septation in humans starts in the last months of gestation and continues through the perinatal period for at least 1 to 2 years.11,14,15
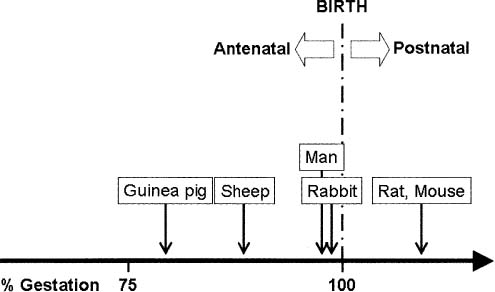
FIGURE 2–6 Onset of alveolarization in various mammalian species.
Regulation of Lung Organogenesis
The organogenesis of the fetal lung and growth and differentiation of the distal lung parenchyma involve complex and highly coordinated processes that result in the formation of gas-exchange units (alveoli). During the past decade, molecular biology and genetic experimental approaches have addressed the regulation of branching morphogenesis and cellular differentiation within the lung.
Techniques such as targeted gene disruption, gene knock-in/knockout, in vitro mutagenesis, and use of antisense oligonucleotides have opened avenues to study specific cellular and tissue functions of genes or their protein products in vivo or in vitro. Studies of the molecular and genetic regulation of lung morphogenesis have focused on two model systems: the airways of the fruit fly Drosophila melanogaster and the mouse lung. Key genes that direct the branching patterns of the airways in these species have been identified and the early steps in respiratory branching delineated. The developmental programs in Drosophila and mice share several important organizational features, suggesting a conserved biologic scheme for pattern formation, branching, and lung morphogenesis.
The common theme emerging from these studies is that lung development proceeds in a coordinated manner from sequential, highly coordinated interactions between epithelium derived from foregut endoderm and the surrounding mesenchyme derived from splanchnic mesoderm. These epithelial-mesenchymal interactions occur through the coordinated spatiotemporal expression of peptide growth factor signaling molecules, transcriptional factors, and extracellular matrix-related molecules. The integration of these molecular inputs instructs organized patterns of cell proliferation, movement, differentiation, and death that determine lung structure and physiologic function. After the establishment of capillary networks in the fetal lung during the later stages of development, systemically derived hormones and other factors also serve important regulatory roles.
Although lung development is a continuum, for the sake of simplicity it can be viewed as occurring in three phases. The first phase involves specification of the lung primordium, septation from the esophagus, and formation of the trachea. The second phase involves branching morphogenesis. The third phase consists of alveologenesis and differentiation of distal epithelial cell types. These three phases are controlled by complex, partially overlapping molecular mechanisms. The following brief discussion of the current knowledge of the mechanisms controlling lung bud formation, branching morphogenesis and alveolarization, and vascular development is inevitably incomplete. The reader is referred to several excellent reviews.1,37–42
It needs to be emphasized that several of these molecular signaling pathways that are critical during normal lung development are also important in the mature lung in conditions such as injury and repair, and carcinogenesis. Furthermore, dysregulation of the signaling pathways may underlie specific congenital malformations of lungs and airways.
Initiation of Lung Morphogenesis
Around embryonal day 25 in humans, epithelial buds sprout ventrally from the foregut into the surrounding mesenchyme to form the trachea and left and right primary bronchi. Current knowledge about the molecular regulation of specification of the lung primordium is limited. Recent studies have implicated members of the winged-helix hepatocyte nuclear factor (HNF)-3 family of transcription factors and a sonic hedgehog (Shh)-independent glial nuclear protein (Gli) pathway in the temporal and spatial determination of the lung bud.42–44
Tracheoesophageal septation and tracheal morphogenesis are believed to be instructed by the homeodomain transcriptional factor Nkx2.1 [also known as thyroid transcription factor (TTF)-1] and retinoids, as well as the interaction of sonic hedgehog and the Gli family of transcription factors (reviewed elsewhere42). The secreted morphogen Shh, which is widely produced by the foregut endoderm, is upregulated specifically in distal regions where branching occurs.1,45,46 Shh from epithelial cells binds to the patched (ptc)/smoothened (smo) receptor complex on adjacent mesenchymal cells, resulting in activation of Gli family transcription factors.37,43 Shh is believed to play a critical role in epithelial patterning by stimulating mesenchymal proliferation and regulating production of fibroblast growth factor (FGF)-10.37,47
Shh null mutant mice exhibit grossly abnormal foregut development.47 These mice also lack the dorsoventral separation of the esophagus and trachea. Failure of branching and growth after the formation of the primary lung buds leads to formation of bilateral rudimentary sacs that arise from a single tracheoesophageal tube.44,47 Interestingly, the normal proximodistal differentiation of epithelial cells is preserved, indicating that Shh is essential for tracheoesophageal septal formation, branching morphogenesis, and lobation, but not for differentiation of the pulmonary epithelium.
Branching Morphogenesis
The mammalian lung—like the Drosophila tracheal system—develops by sequential rounds of branching. The primary bronchi elongate and branch, sprout secondary bronchi, which then sprout tertiary bronchi, and so on. Branching continues for about 20 to 22 generations in humans, yielding an estimated 17 million branches of the human lung. In humans, bronchial branching is highly stereotyped until at least 16 generations of the respiratory tree, indicating that the early stages of branching are under rigid developmental control. From 16 to 23 airway generations, the last of which leads into the alveoli, branching appears to follow a more heterotypic, possibly stochastic, distribution.
Epithelial-Mesenchymal Interactions
Morphogenesis of mammalian lungs depends on precisely controlled paracrine signaling between the endodermal epithelium and the surrounding mesenchyme.48 Embryologic tissue recombination experiments have demonstrated that the presence of mesenchyme is necessary and sufficient to initiate lung budding and that the central mesenchyme surrounding the proximal airways has a different inductive potential from that surrounding the distal lung bud.49–53 These latter observations suggest that the mesenchyme might contain spatially restricted cues that direct epithelial branching. Tissue recombination experiments using porous filter separation have demonstrated that induction of early embryonic lung branching morphogenesis can be achieved even in the absence of mesenchymal contact, as long as soluble mesenchymal factors are present.54–56
Just as mesenchymal signaling has an inductive effect on epithelial branching morphogenesis, retrograde signals from the branching epithelium contribute to patterning, growth, and differentiation of the surrounding mesenchyme. Mice carrying a dominant-negative mutation in the epithelial FGF-10 receptor (a potent inducer of branching morphogenesis) lack mesenchymal structures such as pulmonary blood vessels.57 Shh has also been implicated in retrograde signaling of mesenchymal differentiation.58
Peptide Growth Factors
A variety of peptide growth factors and their cognate receptors play critical inductive or permissive roles in pattern formation, cell proliferation, and differentiation of the developing lung via mesenchymal-epithelial interactions. Peptide growth factors and cognate receptors expressed in the mouse embryonic lung, include fibroblast growth factors (FGFs), epidermal growth factors (EGFs), hepatocyte growth factor (HGF), insulin-like growth factor (IGF), keratinocyte growth factor (KGF), platelet-derived growth factor (PDGF), and transforming growth factor-β3 (TGF-β3). Their effects on lung development have been determined by complementary gain versus loss-of-function experiments in early embryonic mouse lung organ culture, and transgenic and null mutant mice.48,59–68
Fibroblast Growth Factors
FGFs are a complex and continuously expanding family of growth factors consisting of at least 18 ligands that signal through four cognate tyrosine kinase FGF receptors. Ligand-receptor interactions activate a variety of intracellular signaling cascades that regulate cell proliferation, differentiation, and patterning in several developing systems.1,69–71
Accumulated evidence suggests that FGF family members serve an important role as mediators of the mesenchymal-epithelial interactions essential for lung bud formation, branching morphogenesis, and cellular differentiation.49,72 FGF-10 is believed to play a key role in lung patterning and branching morphogenesis through binding to the FGF type 2 receptor (FGFR-2).73,74 In the early stages of lung development, FGFR-2 is expressed along the entire proximal-distal axis of the respiratory tree.75,76 In contrast, the FGF-10 ligand is expressed in a complex and dynamic pattern at discrete sites in surrounding mesenchyme at prospective sites of primary, secondary and tertiary branch formation.1,38,73,74 FGF-10 may act as chemoattractant for epithelial lung buds.73,74
Mice homozygous for a targeted deletion in the FGF-10 gene have a trachea, but no bronchi or lungs,77,78 implicating FGF-10 as an essential regulator of bronchial formation and branching morphogenesis. Inhibition of bronchial branching is also observed in mice expressing a dominant-negative form of the FGFR-2 receptor in the pulmonary epithelium.63,79
In addition to directing early bronchial branching, the FGF-10/FGFR-2 axis directs successive rounds of branching. At each stage of branching, the FGF effector pathway is modified by specific genetic feedback controls and other signals to produce distinct branching patterns. As buds extend toward the FGF-10 signaling centers in the mesenchyme, outgrowth of the budding tip is regulated by differential expression of bone morphogenetic protein (BMP)-438,80,81 and TGF-β180,82 (Fig. 2–7). The Drosophila sprouty homologues, mspry 2 and mspry 4, that are expressed in the epithelium and mesenchyme, respectively, inhibit FGF-10 signaling and limit branch formation.83–85
Keratinocyte growth factor (KGF, FGF-7) is a member of the FGF family that has many similarities with FGF-10.86 The gene sequences have significant homology, and both peptides are expressed by mesenchymal cells and share a receptor (KGFR, FGFR-2-IIIb) that is expressed exclusively on epithelial cells.86 A role for KGF in lung development was first suggested by the observation that its receptor, FGFR-2, is expressed in developing lung.75 Subsequently, targeting a dominant negative FGFR-2 to the lung was found to result in inhibition of branching morphogenesis and epithelial differentiation,63 whereas overexpression of KGF in the mouse lung epithelium resulted in a cystadenoma-like pulmonary malformation.87,88 These studies, and additional studies in explanted rat lungs,67,89 suggest a role for KGF in normal lung morphogenesis. Surprisingly, however, KGF null mice have histologically normal lung development and survival.90 The lack of an effect of the KGF null mutation on lung development may be partially explained by the overlapping function of FGF-10 and KGF in the regulation of branching morphogenesis.86
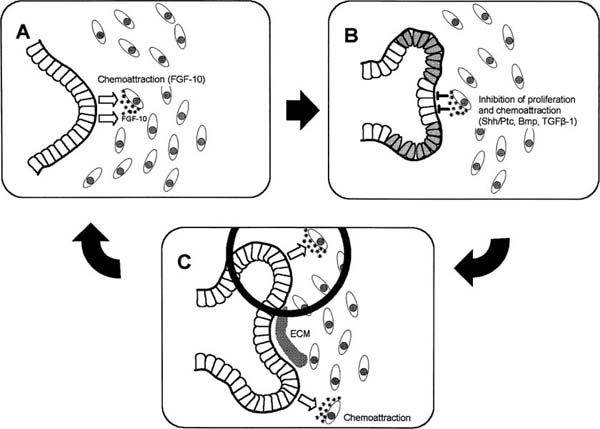
FIGURE 2–7 Schematic model for reiterating bud formation during branching morphogenesis in the developing lung. Local expression of fibroblast growth factor-10 (FGF-10) in the mesenchyme induces directional bud growth. As the bud approaches the chemotactic source of FGF-10, further growth of the tip of the epithelial bud is inhibited by a negative feedback mechanism between the epithelial cells and FGF-10–expressing mesenchymal cells. FGF-10–induced increase in bone morphogenetic protein (BMP)-4 levels in the tip cells and subepithelial transforming growth factor-β1 (TGF-β1) expression lead to inhibition of proliferation. Chemoattraction is inhibited when FGF-10 expression is downregulated by high levels of Shh/Ptc at the tip of the growing bud. Inhibition of cell proliferation and chemoattraction limit bud outgrowth, resulting in cleft formation. As the initial chemotactic center is dampened, FGF-10 expression is relocalized laterally, leading to two new sources of the chemoattractant. The processes described in A and B are reiterated, and two new distal branches form. At the site of cleft formation, TGF-β1 induces the synthesis of extracellular matrix components along the epithelialmesenchymal interface to prevent further budding. (Modified from Lebeche D, Malpel S, Cardoso WV. Fibroblast growth factor interactions in the developing lung. Mech Dev 1999;86:125–136; and Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 1997;124:4867–4878)
In addition to its role in lung morphogenesis, KGF has important effects on epithelial differentiation in the developing lung.55,76,91–93 Furthermore, like FGF-10, KGF plays an important role in fetal lung fluid secretion, a process that is closely linked to lung morphogenesis.94,95
The functional role of other FGFs in lung morphogenesis is less certain. During postnatal life, FGFR-3 and FGFR-4 play an essential role in regulating alveolarization.96 Double null mutation of FGFR-3 and -4 results in a postnatal lethal pulmonary phenotype characterized by excess elastin deposition and failure of alveolar development.96
Epidermal Growth Factors
EGF, TGF-α, and amphiregulin are EGF family growth factors that act through a common receptor (EGFR).62,97 All three ligands are produced by the mesenchyme and influence mouse lung branching morphogenesis and cytodifferentiation via binding to the epithelial EGFR.62,97 Targeted deletion of EGFR in mice has variable effects on lung morphogenesis depending on the genetic background.60,65,98,99
Insulin-Like Growth Factors
The insulin-like growth factors (IGFs) and their binding proteins and receptors are expressed in human and rodent lung throughout gestation.100–104 Although the distribution and levels of IGF-I and -II and their receptors do not appear to change during gestation, the IGF-binding proteins (IGFBPs) 1 to 6 are developmentally regulated, implicating the IGFBPs in mediating spatiotemporal IGF signaling.103 IGF-I receptor null mice die at birth of respiratory failure. The lungs are hypoplastic but branching morphogenesis appears to be preserved.105 IGF-II receptor null mice, which die after birth due to cardiac anomalies, have abnormal alveoli but no apparent branching defects.106
Hepatocyte Growth Factor
Hepatocyte growth factor/scatter factor (HGF) and its receptor, c-met (a tyrosine kinase), are expressed in many developing organs, including the lung.86 In the primitive lung, HGF expression is restricted to the mesenchyme, whereas receptor expression is confined to the epithelium, suggestive of inductive epithelialmesenchymal interactions.107,108 Targeted deletion of HGF is lethal in mouse embryos, but lung branching appears to be normal.109 In rat fetal lung explants, exogenous HGF stimulates branching morphogenesis,110 whereas antisense HGF oligonucleotides in embryonic rat lung organoids block alveolar and bronchial morphogenesis.111 However, unlike KGF or acidic FGF, HGF alone is insufficient to restore branching morphogenesis of fetal lung epithelial explants in the absence of mesenchyme.110 HGF thus appears to play a permissive but not essential role in branching morphogenesis in the lung, perhaps due to redundancy of parallel mesenchymal-epithelial signaling pathways.
Platelet-Derived Growth Factor
Platelet-derived growth factor (PDGF) peptides are dimeric molecules composed of two peptide chains (A and B), which can bind as hetero- or homodimers to the PDGF tyrosine kinase receptors. The PDGF-AA and PDGF-BB homodimers and PDGF receptor are expressed in fetal rat lung, where expression of the PDGFs is restricted to the epithelium, and expression of cognate receptors is localized to the mesenchyme.112 PDGFAA regulates DNA synthesis and branching in early mouse embryonic epithelium.66 PDGF-A homozygous null mice are lethal either in utero before E10 or postnatally. PDGF-A null mice that survive until after birth show hyperinflated, emphysema-like alveoli due to defects in septation.113 Although it plays a role in branching morphogenesis, PDGF-A is believed to be most critical in alveologenesis by regulating proliferation and migration of smooth muscle cells surrounding developing alveoli.
Transforming Growth Factor-β
The TGF-β1, 2, and 3 peptides and TGF-β type I and type II receptors are differentially distributed in embryonic lung.114–117 Both TGF-β1, expressed in the mesenchyme, and TGF-β2, expressed in airway epithelium, inhibit pulmonary branching morphogenesis117,118 (Fig. 2–7). These actions are mediated through the TGF-βIIR, a serine/threonine kinase.119 Smad family proteins function as downstream mediators of TGF-β signaling.120,121
By contrast, TGF-β3 appears to have an essential stimulating role in lung morphogenesis. Mice homozygous for a targeted deletion of the gene encoding TGF-β3 exhibit delayed pulmonary development and decreased surfactant protein C (SP-C) expression.64 Interestingly, TGF-β3 gene expression in fetal lung fibroblasts is strongly induced by corticosteroids,122 suggesting that the maturation-inducing effects of corticosteroids may be mediated, at least in part, by stimulating TGF-β3 gene expression.
The bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily that also play a key role in lung development, specifically the control of proximal-distal patterning and branching morphogenesis.123,124 BMP-4 is highly expressed in the epithelial cells located at the distal ends of lung buds and less so in the surrounding mesenchyme (Fig. 2–7).45 BMP-4 specifically inhibits proliferation of epithelial cells at the tips of the end buds, thereby facilitating the outgrowth of lateral branches.123 Misexpression of BMP-4 driven by the SP-C promoter in transgenic mice results in small lungs with grossly distended terminal sacs, reduced numbers of type II cells, and decreased epithelial cell proliferation.123
Transcription Factors and Signaling Molecules
In recent years, knowledge of the transcription factors that regulate formation and differentiation of the developing lung has advanced significantly.125,126 Morphoregulatory transcription factors provide a link between epithelial-mesenchymal growth factor signaling and cellular processes such as cell proliferation, migration, differentiation, and death. Although none of the transcription factors participating in lung development are exclusively lung-specific, their combinations and interactions provide the basis for lung morphogenesis and developmental physiology.42
Nkx2.1 (Thyroid Transcription Factor,TTF-1)
The homeodomain protein Nkx2.1 is expressed in cells of the primitive lung bud epithelium and in differentiating epithelial cells. Later in gestation, Nkx2.1 expression is restricted to the distal epithelium and postnatally, it is particularly abundant in alveolar epithelial type II cells.127 Nkx2.1 is essential for the complete induction of embryonic lung branching morphogenesis,128–130 epithelial proliferation, and the development of distal structures, including alveolar epithelial type II cells. Nkx2.1 has a regulatory role in expression of the lung-specific genes SP-A, SP-B, SP-C, and Clara cell secretory protein (CCSP).125
Absence of Nkx2.1 results in a failure of septation of the anterior foregut along the dorsoventral axis,129 similar to that observed in mice lacking genes encoding retinoic acid receptors (RAR)-α1/-β2, Gli2, or Shh. Furthermore, Nkx2.1 ablation results in arrest of lung branching morphogenesis and epithelial cell lineage determination.129 Deletion in the Nkx2.1 gene has been reported in a human infant with both thyroid dysfunction and respiratory failure.131 Nkx2.1 also is decreased in atelectatic areas of the lungs in respiratory distress syndrome (RDS) and in collapsed airways in bronchopulmonary dysplasia (BPD).132
Drosophila Forkhead Homologue Hepatocyte Nuclear Factor Family Proteins HNF-3β and HFH4
The HNF family transcription factors, mammalian homologues of the forkhead family in Drosophila, are believed to determine pulmonary epithelial cell lineage fates, possibly in cooperation with the Nkx2.1 family.37,133–136 HNF-3β is expressed in gut endoderm from the earliest stages of its development. In developing lung, HNF-3β is expressed at higher levels in epithelial cells lining the proximal airways than in cells of the distal airways.135 HNF-3β null mice display a severe embryonic lethal phenotype, in which the primitive foregut fails to close.137 Overexpression of HNF-3β targeted to distal lung epithelium results in arrest of branching morphogenesis, epithelial cytodifferentiation, and vasculogenesis, leading to perinatal death.135
Pulmonary expression of HNF/forkhead homologue 4 (HFH-4), another member of the winged helix family of transcription factors, is restricted to ciliated cells of bronchial and bronchiolar epithelium.138 In HFH-4 null mice, there is an absence of ciliated cells in all tissues, including the respiratory tract, suggesting a role for HFH-4 in ciliogenesis.138
GATA Family Transcription Factors
GATA-6, a member of the GATA family of zinc finger transcription factors, has been implicated in determining epithelial and smooth muscle cell lineage diversity in the lung.37,139 GATA-6 induces differentiation of primitive foregut endoderm into respiratory epithelial cell lineages and is believed to interact with HNF-4, HNF-3β, Nkx2.1, and other GATA family members.37,140 GATA-6 is required for maturation of the gas-exchange area in late gestation.141,142 GATA-5 is expressed in primitive lung mesenchyme and in bronchial smooth muscle.143
C/EBP Family Transcription Factors
The CCAAT/enhancer binding protein (C/EBP) family of transcription factors belongs to the large family of basic region-leucine zipper (bZIP) transcriptional factors.144,145 C/EBPα, β, and δ are believed to play important roles in adipocyte differentiation146 and are expressed at relatively high levels in lipid-synthesizing tissues, including lung.147 C/EBP null mice show pulmonary immaturity and an increased risk of neonatal death from apparent respiratory distress.148
Homeobox (Hox) Genes
Hox genes code for transcription factors that regulate body-axis patterning and specification of regional identity during embryonic development.149,150 Expression of hox genes is spatiotemporally restricted during embryonic lung branching morphogenesis.151 The spatiotemporal patterns of hox gene expression in early embryonic mouse lung are consistent with roles in determining the proximal-distal orientation of the lung and in branching morphogenesis.152–154 Several hox genes including Hox b-3, b-4, and b-5 have been implicated in branching morphogenesis.155 Hox a5 null mutation results in an early neonatal lethal pheno-type, characterized by tracheal occlusion, respiratory distress and a dramatic decrease in surfactant protein production.156
N-myc
N-myc is a member of a family of proto-oncogenes encoding basic helix-loop-helix-leucine zipper (bHLHLZ) transcription factors that bind to regulatory elements of genes involved in cellular proliferation and differentiation. N-myc expression is restricted to certain epithelial cell lineages of the embryo.157 In developing lung, N-myc expression is high in bronchiolar epithelium at the distal tips of developing airways.158 N-myc null embryos die in utero with major defects in various organs, including lungs.157 An important role for N-myc in branching morphogenesis has been suggested by complete or partial deletion experiments in mice.158,159
Glucocorticoid Receptor
The glucocorticoid receptor (GR) is a member of the nuclear receptor superfamily of transcription factors. Upon binding hormone, GR enters the nucleus and binds to specific glucocorticoid response elements (GREs) in the regulatory regions of target genes. GR expression in developing lung is mainly confined to the mesenchyme.160 Mice homozygous for a targeted deletion of the GR gene die soon after birth due to pulmonary underdevelopment.161
Extracellular Matrix Components
Extracellular matrix (ECM) formation is an essential component of lung development. The ECM can serve as a structural support for organ morphogenesis, provide signaling cues that regulate cell behavior, and modulate growth factor availability or activity.162
A variety of ECM components contribute to pulmonary tissue growth and morphogenesis, although much remains to be learned about their specific roles.163–166 ECM components of the basement membrane including laminins, entactin/nidogen, collagen type IV, perlecan, secreted protein ascistic and rich in cysteine (SPARC), and fibromodulin, play an important role in the regulation of epithelial-mesenchymal and cell–matrix interactions during lung morphogenesis. These ECM components not only support the tissue architecture, but also are believed to play an active role in the modulation of cell proliferation and differentiation.167 The role of the ECM in fetal lung remodeling has been reviewed.37
The development of the ECM may be as critical to alveolarization as it is to differentiation and morphogenesis of the lung throughout earlier fetal life. Synthesis and accumulation of collagen,168 proteoglycan,169 and elastin170,171 peak by 80% completion of gestation, synchronous with the onset of alveolarization. Modulation of elastin structure or synthesis causes defective alveolarization and interferes with mechanical properties of the lung including loss of elastic recoil.172
During lung development, the ECM undergoes dynamic proteolytic remodeling. Matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of MMPs (TIMPs), play an important role in the regulation of ECM remodeling and are important in the morphogenesis of fetal lungs.173,174 Altered levels of specific MMPs or TIMPs have been identified in children with lung disease.175,176
Alveologenesis
Although recent advances in molecular biology have led to significant progress in our understanding of the regulation of lung organogenesis, the regulation of alveolarization is only now beginning to be elucidated.
FGF, PDGF, and EGF signaling pathways have been implicated in alveologenesis. During postnatal life, FGFR-3 and FGFR-4, receptors that bind several of the FGFs, cooperate to regulate alveolarization. A double mutation of these genes results in a postnatal lethal pulmonary phenotype in which there is failure of alveolar septation, resulting in an emphysematous appearance.96 Mice lacking the growth factor PDGF-A also exhibit a defect in pulmonary alveologenesis, apparently caused by a loss of alveolar smooth-muscle myofibroblasts and parenchymal elastic fibers.113
Neonatal EGF receptor-deficient mice demonstrate a defect in alveolarization, associated with impaired branching morphogenesis.99 Inactivation of the EGF receptor affects type II cell maturity, with decreased expression of the distal epithelial marker SP-C. In contrast, exogenous EGF accelerates alveolar type II cell differentiation in fetal lungs.177 Retinoids and glucocorticoids affect alveologenesis, but their precise roles remain undetermined [reviewed in Minoo42].
Alveolar Type II Cell Differentiation
Pulmonary surfactant is a developmentally and hormonally regulated macromolecular complex composed primarily of lipids and proteins. Surfactant is synthesized by alveolar epithelial type II cells and stored in characteristic lamellated intracellular inclusions called lamellar bodies. After secretion, the lamellar bodies undergo a structural transformation into highly organized structures called tubular myelin, which gives rise to a monolayer surface film of surfactant lipids and proteins. The phospholipid layer acts to reduce surface tension in the alveolus, thereby stabilizing the alveoli and preventing collapse at end-expiration.
Lung surfactant contains four associated proteins, SP-A, SP-B, SP-C, and SP-D. SP-B and SP-C, extremely hydrophobic proteins, are critical in conferring surfactant-like properties on the phospholipids present in the alveolus.178 SP-A and SP-D are members of the collectin subgroup of the C-type lectin family of proteins179 and appear to function in immune defense and processing of secreted surfactant, respectively (reviewed elsewhere125).
Surfactant protein gene expression is cell-specific and developmentally regulated.125 SP-A gene expression occurs primarily in alveolar type II cells and nonciliated bronchioalveolar (Clara) cells of the bronchioles.180,181 However, in human midtrimester fetal lungs, SP-A has also been localized to tracheal and bronchial glands and conducting airway epithelium.182 SP-A gene transcription is initiated in fetal lung after 75% of gestation is completed and is maximal immediately before birth. In humans, immunoreactive SP-A protein is detecTable in amniotic fluid at 30 weeks’ gestation and increases further during development, concomitantly with an increase in the lecithin-sphingomyelin (L/S) ratio.183
SP-B messenger RNA (mRNA) transcripts are present in type II cells and bronchiolar epithelial cells,180 whereas SP-C gene expression is restricted to type II cells.184,185 SP-B and SP-C gene expression is initiated at an earlier stage in fetal development than SP-A. In human fetal lung, mRNAs for SP-B and SP-C are detectable from 13 weeks’ gestation onward.186 Their levels continue to increase during gestation so that by 24 weeks SP-B and SP-C mRNA levels are 50% and 15% of adult levels, respectively.186
SP-D is expressed in nonciliated bronchioloalveolar epithelial and type II cells and tracheobronchial glands of the lung, as well as in several nonpulmonary glandular tissues.187 In human lungs, SP-D mRNA is first detected at low levels in the second trimester; expression continues to increase throughout gestation and postnatally.188 The different temporal patterns of expression indicate that the genes encoding the surfactant-associated proteins are independently regulated during development. Surfactant protein gene expression in fetal lungs is regulated by several hormones and other factors, including retinoids, insulin, growth factors, cytokines, glucocorticoids, and cyclic adenosine monophosphate (cAMP).125
Specification of Left-Right Asymmetry
Despite initial symmetry during the sprouting of the lung buds, the left and right lungs develop asymmetrically. Several regulatory genes, including members of the TGF-β superfamily (lefty-1, lefty-2, nodal, gdf1), Shh, Pitx2 (a homeobox gene), and forkhead homologue of HNF (HFH-4), are involved in the left-right asymmetry of the murine lung.138,189–191 Lefty-1 null mutant mice show a variety of left-right isomerisms in visceral organs including bilateral “left” lungs.192 Left-right orientation is also dependent on left-right dynein (lrd), a microtubule motor protein.192 Targeted deletion of the murine forkhead family gene, HNF-4, results in the absence of left-right dynein expression and, hence, of cilia in association with random left-right laterality of visceral organs, including the lungs.138
Apoptosis in Lung Development
Apoptosis is a distinct form of active programmed cell death characterized by coordinated loss of cell function and rapid biochemical and morphologic changes, culminating in cell death without associated inflammation.193 In view of the well-established role of apoptosis in developmental modeling processes involving other organ systems,194–197 apoptosis is believed to play a role in fetal lung development as well. Although its role and regulation are far from understood, recent research implicates apoptosis as a key player in both organogenesis and alveolar remodeling of the developing lung (reviewed elsewhere198).
Apoptotic activity has been observed during all stages of lung development. During the embryonal stage, apoptotic cells are almost exclusively found in the peripheral mesenchyme surrounding branch points or newly formed buds, representing the sites of active epithelial branching morphogenesis and mesenchymal remodeling.199,200 During the early stages of fetal development, much of the apoptotic activity remains localized to peribronchial and perivascular mesenchymal cells, likely representing the final stage of branching morphogenesis.201–204 In postcanalicular lungs, apoptotic activity gradually shifts to alveolar epithelial cells and interstitial mesenchymal cells.202–204 After birth, a surge of apoptosis is observed in alveolar epithelial type II cells and interstitial fibroblasts.172,202,205,206 Fibroblast apoptosis during and after alveolarization likely plays an important role in the thinning and involution of the alveolar septum and contributes to attenuation of the gas–blood barrier.
The signaling pathways mediating developmental lung apoptosis remain to be elucidated. One candidate regulator of alveolar epithelial apoptosis is the Fas/Fas-ligand (FasL) death signaling system. The Fas/FasL system is a widely distributed signal transduction pathway in which ligand-receptor interaction triggers cell death.207 Fas (CD95/APO1), a member of the tumor necrosis factor/nerve growth factor receptor superfamily, is activated by binding to FasL, stimulating downstream activation of intracellular caspases leading to apoptosis. Fas and FasL expression in fetal and adult lungs has been localized to alveolar epithelial type II cells and bronchial epithelial Clara cells.204,208–211 In fetal rabbit lungs, apoptotic remodeling during the transition from the canalicular to the saccular stage coincides precisely with a dramatic upregulation of FasL expression.204 Although a well-defined spatiotemporal pattern of Fas/FasL signaling in developing lungs seems to implicate the Fas/FasL system as a pivotal mediator of postcanalicular type II cell apoptosis, many questions remain unresolved, such as the apparently normal lung development of mice deficient in Fas, FasL, or critical downstream Fas/FasL signaling molecules.212 Furthermore, although Fas/FasL may be important in alveolar epithelial apoptosis, recent evidence suggests that the mitochondrial cytochrome c-dependent pathway may be important in apoptosis of alveolar mesenchymal cells.213
The temporal pattern of lung apoptosis synchronous with important structural alterations implicates apoptosis in pulmonary histogenesis and remodeling. Although the developmental controls of mesenchymal and epithelial lung apoptosis remain to be elucidated, candidate regulators include mechanical distention/stretch, oxygen tension, glucocorticoids, and other hormones. Fetal lungs are exposed to cyclic as well as tonic distention, caused by the combined effects of fetal breathing movements and lung liquid secretion. Similarly, postnatal breathing induces marked airspace distention during the stage of alveolarization. Studies in fetal tracheal occlusion models203 have demonstrated that mechanical distention of fetal rabbit lungs results in a dramatic increase of alveolar epithelial type II cell apoptosis, which likely contributes to the observed type II cell depletion and surfactant deficiency after tracheal occlusion. Similarly, in vitro experiments have demonstrated that stretch can induce apoptosis in adult alveolar epithelial type II cells214 and fetal fibroblasts.213
Regulation of Postglandular Distal Lung Remodeling
Postglandular lung development comprises the canalicular, saccular, and alveolar stages and results in the formation of the gas-exchange surface of the lung by means of septation, maturation of epithelial and mesenchymal cells, and development of the alveolar capillary system.36,215 Lung development during these stages is highly susceptible to various local and systemic signals, which include genetic factors, oxygen tension, nutrition, and hormones. Complex relationships exist between these humoral, hormonal, and physical forces acting on the developing lung. Although many of the molecular mechanisms underlying the effects of these regulators on lung growth remain to be delineated, it is likely that these intrinsic and extrinsic modulators exert their effects via specific developmentally regulated and evolutionary conserved molecular pathways.
Hormones
Glucocorticoids
In the late 1960s Liggins216 was among the first to demonstrate that cortisol treatment of fetal lambs accelerates lung maturation. Subsequently, Liggins and Howie217 reported that antenatal corticosteroids given to mothers at risk of preterm delivery can decrease the incidence of RDS in preterm newborns. Since these initial reports, numerous studies have demonstrated the effects of glucocorticoids on surfactant production and trafficking both in vivo and in vitro.218,219
Glucocorticoids have been shown in several different model systems to accelerate physiologic, morphologic, biochemical, and molecular indices of lung maturation and to induce all components of the surfactant system.186,188,216,218,220–229 Glucocorticoids also have effects on structural lung growth and development,230–234 on antioxidant enzymes,232,235–238 on lung tissue growth factors,239,240 on inflammatory mediators, and on the regulation of pulmonary fluid absorption.232,241–243 Finally, glucocorticoids increase the number of β-adrenergic receptors in the lung,244,245 which may facilitate the transition of the lung to air breathing.246
In addition to the maturational effects of exogenous glucocorticoids, experimental data support a physiologic role for endogenous glucocorticoids.247 In most mammalian species, including humans, free corticosteroid concentrations rise substantially during the last 10% of gestation, synchronous with a surge in surfactant synthesis and accumulation.218 Surgical or biochemical glucocorticoid blockade delays the development of the surfactant system and impairs the structural development of the fetal lung.248–251 The late gestational rise in fetal cortisol production is associated with increased expression of the glucocorticoid receptor (GR) in pulmonary tissues,218,252–259 suggesting that the control of lung maturation by glucocorticoids may be regulated in part by developmental modulation of GR availability. GR null mice die soon after birth as a result of pulmonary underdevelopment,161 emphasizing the importance of GR in lung development.
The acceleration of normal lung development by glucocorticoid treatment has led to widespread clinical use of ante- and postnatal glucocorticoids aimed at accelerating lung development in preterm infants. Antenatal glucocorticoids (dexamethasone and betamethasone) are frequently administered during premature labor,260,261 ideally between 24 hours and 7 days before the infant is born.262 The effects of antenatal glucocorticoid therapy include a reduction in the severity of RDS, reductions in the incidence of other complications of prematurity (such as intracranial hemorrhage and necrotizing enterocolitis), and a reduction in neonatal mortality.
The potential long-term adverse effects of repeated perinatal glucocorticoid administration, particularly with respect to fetal growth, long-term pituitary-adrenal function, and neurodevelopmental outcome, have become the focus of clinical and experimental attention263 (reviewed elsewhere264,265). The use of postnatal pharmacologic glucocorticoid administration in particular is the subject of ongoing controversy. Currently available evidence suggests that postnatal glucocorticoids in some circumstances may be associated with a reduction in the incidence and severity of chronic lung disease and a reduction in neonatal mortality (reviewed elsewhere264). However, adverse effects are frequent and include gastrointestinal bleeding, hypertension, hyperglycemia, and adrenal suppression. In addition, there are increasing concerns that dexamethasone may permanently alter the structure and function of certain tissues following postnatal administration, especially lungs and brain.266–269
Thyroid and Parathyroid Hormones
There is evidence that thyroid hormones are involved in some aspects of lung maturation, namely, surfactant production and the development of antioxidant enzyme systems (reviewed elsewhere270–273). Thyroid hormone may also be a physiologic regulator of septation late in gestation and early postpartum.274,275 Although some studies have shown that administration of thyroxine (T4) or triiodothyronine (T3) alone can accelerate alveolar epithelial cell differentiation,274–276 others have suggested that thyroid hormones alone have little effect on surfactant synthesis; rather, they act in an additive or synergistic fashion with glucocorticoids to accelerate lung maturation.218,277–280 Recent evidence suggests thyroid hormone may also affect early lung morphogenesis. Exogenous T3 treatment of murine embryonic explants during early lung development induces accelerated epithelial and mesenchymal cell differentiation at the expense of premature arrest of branch formation and lung growth.281
Parathyroid hormone (PTH) receptors have been localized to lung.282,283 PTH and PTH-related peptide (PTHrP) bind to a common receptor.284 PTH and PTHrP can stimulate phosphatidylcholine synthesis in fetal lung explants and in cocultures of pulmonary fibroblasts with type II pneumocytes,285 suggesting that these proteins can act directly on lung tissue and may be involved in the regulation of lung maturation.
Catecholamines
Birth is associated with a surge in catecholamine release from the adrenal medulla.286 Endogenous catecholamines are believed to play a major role in the adaptation to extrauterine life. Catecholamines contribute to the postnatal dilatation of both pulmonary airways and arteries, thus ensuring optimal aeration of the lung and a sustained fall in pulmonary vascular resistance. Endogenous catecholamines also play a role in the clearance of fetal lung fluid at birth, in concert with other maturational hormones such as glucocorticoids and thyroid hormones.287 Finally, autonomic transmitters such as epinephrine contribute to the maturation of the surfactant system.288,289
Adrenergic agonists act on pulmonary smooth muscle cells and type II cells through β-adrenergic receptors (βARs), which activate adenyl cyclase via a stimulatory G protein to cause an increase in cAMP.290,291 During development, the number of βARs increases in pulmonary arterial and airway smooth muscle cells.292
Androgens
During late gestation, male fetuses exhibit a delay in lung structural maturation and surfactant synthesis.293 This sexual dimorphism involves both structural and functional aspects of mid- and late-gestation lung development. Male fetuses have delays in formation of the thin air–blood barrier, appearance of foot processes, cell–cell contacts, and lamellar bodies and loss of basement membrane continuity.294,295 Functionally, males have delayed surfactant synthesis and reduced compliance and functional residual capacity.296–298 These differences are thought to account for the increased incidence of respiratory distress syndrome among male premature infants compared with female infants.299,300
The delay in surfactant synthesis in male fetuses has been attributed to higher androgen concentrations.301 Clinical and experimental data indicate that androgens may have an inhibitory effect on endogenous and glucocorticoid-stimulated surfactant synthesis.218 Treatment with dihydrotestosterone (DHT) during sexual differentiation inhibits surfactant phospholipid production in the female fetus, whereas treatment with the antiandrogen flutamide increases surfactant phospholipid production in the male fetus.301 In addition to androgens, müllerian inhibiting substance (MIS), a Sertoli cell–derived glycoprotein made early in the ontogeny of the testis, has been shown to inhibit lung maturation.302
Insulin and Diabetes Mellitus
Maternal diabetes is associated with delayed fetal pulmonary maturation, as measured by the delayed appearance of biochemical markers of lung maturity such as phosphatidylglycerol and an increased lecithin/sphingomyelin ratio. Delayed fetal lung maturity is believed to account for the excessive neonatal pulmonary morbidity in diabetic pregnancies303 and for the occurrence of hyaline membrane disease even in term gestations.304–313 Despite the longstanding recognition of the detrimental effects of maternal diabetes during pregnancy and the introduction of clinical protocols to minimize its effects, the mechanism of delayed lung maturation in diabetic pregnancies has not been fully elucidated. Chronic maternal hyperglycemia with secondary fetal hyperinsulinemia has been shown to alter fetal lung maturation and function.308,314,315
The risk for development of fetal lung immaturity and hyaline membrane disease in diabetic pregnancies is linked to maternal glucose control.311,316 The rate of immature fetal lung profiles at amniocentesis is higher in pregnant women with poorly controlled diabetes than in pregnant women without diabetes.311 Strict glucose control may lower the risk of fetal pulmonary immaturity to that seen in the nondiabetic population.311,317 There appears to be no difference between gestational and pregestational diabetic pregnancies.311 In addition to delayed structural and functional maturation, preterm infants of diabetic mothers have been reported to exhibit increased muscularization of small pulmonary arteries and neonatal pulmonary hypertension.318
Retinoids
Vitamin A (retinol) and its biologically active metabolite, retinoic acid, have well-established roles in the development of both the conducting airways and alveolar portions of the lungs.319–321 These effects are mediated by retinoid receptors, members of the nuclear receptor superfamily that regulate the expression of target genes. Retinoids have a critical role in early branching morphogenesis of the lung. The similarities of defects in lung morphogenesis between retinoic acid receptor (RAR) RAR-α1/RAR-β2 double knockout mice and Shh/-and Gli2-/- Gli3+/− mice have suggested an interrelationship between the Shh/Gli and retinoid signaling pathways in early lung development.125,322
Furthermore, evidence has accumulated that retinoids are important regulators of alveolus formation (reviewed elsewhere323). Exogenous all-trans retinoic acid (ATRA) induces the formation of additional alveoli in newborn rats,324 whereas retinoic acid receptor null mice display a marked failure of alveolar septation.325 Studies of adult mice with genetic failure of lung septation,326 rats with glucocorticoid-inhibited alveolarization,324,326 rat pups with hyperoxia-inhibited lung septation,327 and mice with elastase-induced emphysema328,329 have also shown that retinoids promote alveolar formation beyond the normal developmental window for septation.
Retinoic acid affects gene expression of over 300 gene products330 and acts as an antioxidant. There is evidence that retinoids may exert their primary effects on the lung through modulation of genes involved in morphogenesis319,320,331–333 and in extracellular matrix synthesis.325,330 Retinoic acid receptor and binding protein expression in lung peaks during the period of most active lung septation.321,334 One of the extracellular matrix components most critical in the process of lung septation and alveolarization is elastin.233 Retinoids upregulate elastin expression in alveolar myofibroblasts in vitro.335 Furthermore, mice carrying deletions of retinoic acid receptors demonstrate decreased lung elastin and impaired alveolarization,325 suggesting a link among retinoic acid, elastin, and lung alveolar formation.
Clinical studies indicate that vitamin A supplementation may be of benefit to extremely low-birth-weight infants by decreasing their risk of chronic lung disease.336,337 The current knowledge of the effects of retinoids on the lung alveolar formation has recently been reviewed by Massaro and Massaro323 and McGowan.338 Whether retinoids may have therapeutic value in humans with too few alveoli for adequate gas exchange, such as established bronchopulmonary dysplasia in preterm infants or emphysema in adults, remains to be determined.
Physical factors
Amniotic Fluid Volume
The relationship between oligohydramnios and lung hypoplasia has been recognized for many years. Potter339 first described pulmonary hypoplasia in fetuses with renal agenesis. Subsequently, pulmonary hypoplasia has been reported in association with other renal dysplasias340 and in conditions characterized by reduced amniotic fluid in the absence of renal anomalies.341,342 The incidence of hypoplasia with oligohydramnios has been reported at between 9% and 25%343,344 but probably is higher because lesser degrees of pulmonary hypoplasia may remain undiagnosed. The severity of the hypoplasia depends on the gestational age at which oligohydramnios occurs, although this correlation is variable.344 Pulmonary hypoplasia was determined to be the primary cause of death in 22% of patients in a neonatal autopsy series345 with a common finding of prolonged rupture of membranes.
The relationship between premature rupture of membranes (PROM) and pulmonary hypoplasia was examined by Rotschild et al.346 Gestational age at the time of PROM was the best predictor of pulmonary hypoplasia, with no cases occurring when the membranes ruptured after 26 weeks. Several authors have confirmed the association of gestational age at the time of membrane rupture and duration of rupture with the risk of pulmonary hypoplasia.344 The mechanisms by which amniotic fluid volume influences lung growth remain unclear. Pulmonary hypoplasia is likely mediated by several factors acting in concert, including mechanical restriction of the chest wall, interference with fetal breathing, and failure of production of fetal lung liquid.347
Restriction of Thoracic Space
Pulmonary hypoplasia occurs in fetuses with bilateral pleural effusions, chylothorax, and other causes of intrinsic thoracic compression,348,349 as well as extrinsic compression found in abdominal and extraamniotic pregnancies.350,351
Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia (CDH) occurs in 1:2500 live births and is associated with a 30 to 60% mortality rate with significant morbidity among survivors.352–354 CDH can occur as an isolated defect or may have other coexistent congenital anomalies.355 There are different types of CDH, including Bochdalek, Morgagni, and central (septum transversum) diaphragmatic hernia.356 The Bochdalek CDH, which occurs in the posterolateral region of the diaphragm, accounts for ~70% of CDH cases. Morgagni CDH is less common and forms in the anterior retrosternal diaphragm. Central (septum transversum) CDH occurs in the midline of the septum transversum and accounts for 1 to 2% of all cases of CDH.
The lungs of CDH patients are smaller than normal, with a decreased number of airways, a reduced number of alveoli, and reduced surfactant production (Fig. 2–8). The lung is mainly affected on the side of the defect, which is most often left-sided; however, the contralateral lung is also smaller than normal. In addition, there is a decreased number of vascular generations, and an increased thickness of smooth muscle in the pulmonary arterial walls,357,358 thought to be the cause of pulmonary hypertension. The combined effects of pulmonary hypoplasia and pulmonary hypertension result in respiratory distress that is associated with high morbidity and mortality in the neonatal period.
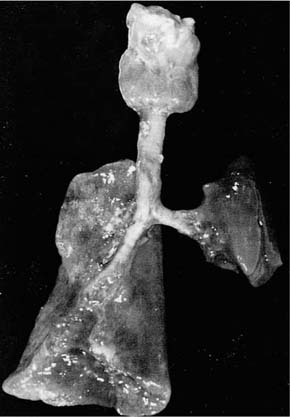
FIGURE 2–8 Left-sided diaphragmatic hernia with severely hypoplastic lung. The right lung was also hypoplastic.
Although CDH has been recognized for nearly 350 years, its pathogenesis and the pathogenesis of CDH-associated lung hypoplasia are not clear (reviewed elsewhere359). Various animal models have been developed to elucidate the mechanisms that underlie the CDH-associated pulmonary hypoplasia. These models include surgical models,360,361 teratogenic (nitrofen) rodent models,362 and genetic models of CDH.363
Pulmonary hypoplasia in association with CDH is traditionally believed to result from direct compression of the lung and inadequate fetal breathing movements due to herniation of abdominal contents into the thoracic cavity. Diaphragmatic defects further alter the mechanics of respiration. Evidence from teratogen-induced rodent models of CDH suggests that defects in diaphragm muscle arise from malformations of the primordial diaphragm, the pleuroperitoneal fold.364 In the nitrofen rodent model, there appears to be concomitant or preceding insults to lung development associated with CDH.365,366 Others have suggested that abnormalities linked with the retinoid signaling pathway early in gestation may contribute to the pathogenesis of CDH.367 A genetic murine model of central (septum transversum) CDH has recently been described.363
Fetal Breathing
Long-term fetal animal experiments have demonstrated that episodic or cyclic breathing movements occur 40% of the time during the last trimester. Although the exact role of this cyclic fetal breathing remains unclear, it is believed that diaphragmatic and chest wall movement provides a stretching stimulus to the lungs, promoting alveolar development. In vitro experiments have demonstrated that mechanical strain can stimulate DNA synthesis and cell division in fetal rat lung cells.368 In contrast, interruption of fetal breathing by spinal cord section or bilateral or unilateral phrenectomy results in arrested lung development.369–372 Pulmonary hypoplasia occurs in neuromuscular affected fetuses such as PenaShokeir syndrome (fetal akinesia sequence).373
Fetal Lung Liquid Production
Structural abnormalities can be produced experimentally by interfering with the normal tracheal egress of fetal lung liquid. Extrapolation from a fetal lung liquid production of 4 to 5 mL/kg/hour in the lamb suggests that human fetal lung liquid production may be 250 to 300 mL/day during the last trimester. This fluid is maintained under a pressure gradient of about 3 to 5 cm H2O across the larynx and results in a lung liquid volume of ~30 mL/kg body weight.374 Periodically the larynx opens to allow egress of amniotic fluid into the amniotic sac. Retrograde movement of fluid during each fetal “breath” is minimal but there is a small, steady movement outward from the trachea. The net flux of fluid from the airway has been measured at ~15 mL/hour and averages about five times higher during fetal breathing periods than during quiescent periods.375
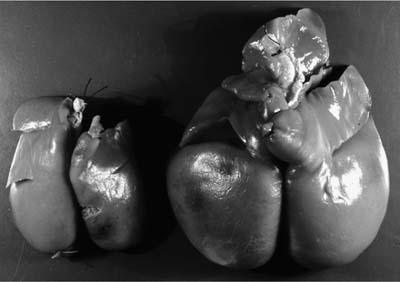
FIGURE 2–9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


