Virtually, all patterns of lung injury have been associated with rheumatologic lung disease, and none is specific for any given entity (Table 50.1). However, many pathologic diagnoses are more common in some connective tissue disorders than others (Table 50.2). It is important, in general terms, for the pathologist to assess acute changes, such as acute alveolar injury and capillaritis, as well as chronic changes such as the degree of fibrosis, as these features have special importance for short- and long-term prognosis, respectively. In cases of diffuse interstitial lung disease, the presence of pleural inflammation and reactive lymphoid follicles favors autoimmune disease, although these may also occur in idiopathic interstitial pneumonias.
Pulmonary arterial hypertension occurs with increased frequency in patients with autoimmune connective tissue disorders (Table 50.3). In this setting, it is often termed “associated” pulmonary arterial hypertension (see Chapters 62 and 63).
There is often difficulty in establishing a specific diagnosis of connective tissue diseases, as autoantibody levels can fluctuate, along with clinical symptoms. Furthermore, it is not uncommon for lung manifestations to occur at the initial phase of disease or even predate the onset of extrapulmonary symptoms. The concept of undifferentiated connective tissue disease has only relatively recently been developed and is a frequent underlying condition in interstitial lung disease, especially nonspecific interstitial pneumonia (Chapter 17).
Because the treatment of autoimmune diseases is immunosuppression, the primary differential diagnostic consideration, both clinically and pathologically, is an infectious process, which must always be excluded by special stains, especially if fibrin or granulomas are present. Drug reactions are also always a diagnostic consideration. Unlike infections, however, there are no specific tests to prove drug toxicity, which is a diagnosis of exclusion and can only be surmised based on clinical correlation.
TABLE 50.1 Pulmonary Pathology in Autoimmune Connective Tissue Disorders
aAn older term, cellular interstitial pneumonia, is not currently in widespread use but describes features of cellular NSIP, Ref.1
Rheumatoid Arthritis
Given the prevalence of rheumatoid arthritis, rheumatoid lung disease is probably the most common “autoimmune lung disorder.” Approximately 10% of patients have clinical lung disease, risk factors being advanced age, severe joint symptoms, and male gender.2 When patients are screened by high-resolution computed tomography, the rate of lung abnormalities is 20% to 44%.3 It has even been suggested that up to two-thirds of rheumatoid arthritis patients will develop interstitial lung disease.4
Clinically, symptoms are variable, including shortness of breath, exercise intolerance, and cough or symptoms related to pleural inflammation. Pulmonary function testing shows obstructive disease, resulting for airway disease; restrictive disease, if there is progression to fibrosis; or a combination of both. There are four major imaging patterns, which correlate to pathologic diagnoses. There are a UIP-like pattern with bilateral subpleural reticulation; an NSIP-like pattern with predominant ground-glass opacities; an inflammatory airway disease pattern with centrilobular branching lines, with or without bronchial dilatation; and an organizing pneumonia pattern with patchy areas of consolidation.5
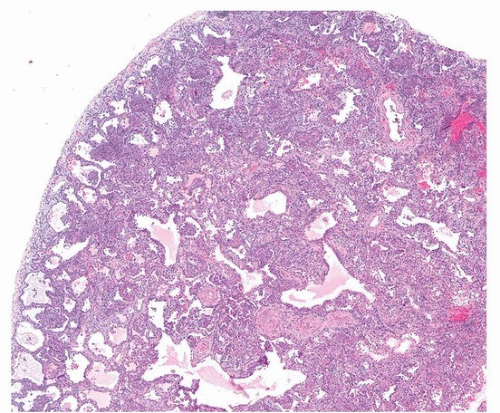
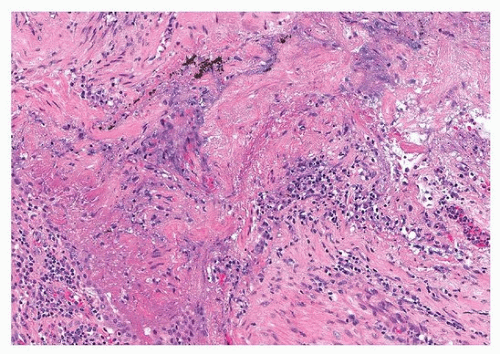
Pathologically, there is a wide range of manifestations of rheumatoid arthritis-interstitial lung disease, although the subacute and chronic phases are most common. Even though organizing pneumonia was classically associated with rheumatoid lung disease, histologic studies show that usual interstitial pneumonia is the most common histopathologic pattern, followed by nonspecific interstitial pneumonia (Fig. 50.1) and then organizing pneumonia.2 The only specific finding is the presence of rheumatoid nodules, which are uncommon in the lung (Fig. 50.2). Caplan syndrome, or rheumatoid pneumoconiosis, is a rare reaction to mineral, coal, or silica dust, manifested by numerous rheumatoid nodules and pulmonary fibrosis in rheumatoid arthritis patients.
Acute lung injury due to diffuse alveolar damage (acute interstitial pneumonia) is an uncommon initial manifestation of rheumatoid lung disease and is similar pathologically and clinically to idiopathic acute interstitial pneumonia. Acute lung injury is more frequently seen as an acute exacerbation of chronic lung disease, sometimes triggered by drug treatment, especially newer immunologic agents.6,7,8 Methotrexate-induced pneumonitis is an uncommon complication with nonspecific clinical and histologic findings and is a diagnosis of exclusion.9
There are a variety of airway diseases that also occur in rheumatoid lung disease. Bronchiolitis, follicular bronchiolitis, and constrictive bronchiolitis have all been described in rheumatoid arthritis patients.10,11 Although some degree of airway obstruction and inflammation is common, constrictive bronchiolitis is rare and has a very poor prognosis. It is difficult to ascertain the exact incidence of constrictive (obliterative) bronchiolitis, because of the confusion between organizing pneumonia (bronchiolitis obliterans-organizing pneumonia), which is common, and true obliterative bronchiolitis.4
Pulmonary vascular disease is a relatively rare manifestation of rheumatoid arthritis. Pulmonary arterial hypertension is more common in other autoimmune diseases (especially systemic sclerosis). Although rheumatoid vasculitis is a relatively common vasculitis, pulmonary involvement is unusual. Small-vessel vasculitis of the lung (capillaritis) is an uncommon cause of diffuse alveolar hemorrhage in patients with rheumatoid arthritis.12
TABLE 50.2 Frequency and Types of Lung Involvement with Autoimmune Connective Tissue Diseases
Connective Tissue Disease
Frequency of Lung Involvement
Phase of Injury
Acute
Subacute
Chronic
Rheumatoid arthritis
20%-40%
Diffuse alveolar damage (uncommon)
Capillaritis (rare)
Follicular bronchiolitis
Organizing pneumonia (common)
UIP (common)
NSIP (less common)
Systemic lupus erythematosus
10%
Capillaritis (rare)
Diffuse alveolar damage
1%-4%
Cellular NSIP
Organizing pneumonia (uncommon)
Lymphoid interstitial pneumonia (uncommon)
NSIP
Lymphoid interstitial pneumonia
3%-8%
Mixed connective tissue disease
50% or more
Acute interstitial pneumonia and capillaritis (rare)
Not described
Nonspecific interstitial pneumonia, with rare honeycombing
50% overall, 20% severe
Systemic sclerosis
70% abnormal lung function
90% abnormal computed tomography scans
Diffuse alveolar damage (uncommon)
Aspiration may occur due to esophageal involvement.
Pleural involvement occurs in almost one-half of patients with systemic lupus erythematosus, and effusions occur in 20%.
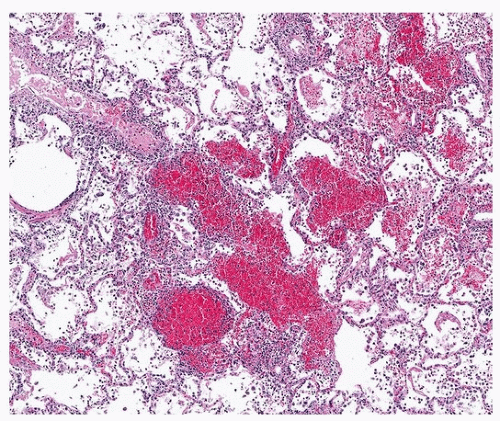
Acute lupus pneumonitis is diagnosed clinically in ˜5% of patients during the course of disease and is manifested by fever, cough, and bibasilar fluffy reticular densities. Acute lung disease is the initial manifestation of lupus in up to 50% of patients.13 There may be an association with anti-dsDNA and anti-Ro/SSA serum autoantibodies. Histologic findings are variable; an early description a half a century ago described a variety of findings, including acute alveolar injury (diffuse alveolar damage), cellular interstitial inflammation (cellular interstitial pneumonia) resembling cellular NSIP, and arteriolar thrombosis.14 Some authors associate acute lupus pneumonitis with diffuse alveolar damage and hyaline membranes only and would include cellular interstitial pneumonia as nonspecific interstitial pneumonia of chronic lupus pneumonitis.1 Capillaritis with pulmonary hemorrhage is also in the spectrum of acute lupus pneumonitis (Fig. 50.3) but is usually clinically designated as diffuse alveolar hemorrhage (as opposed to lupus pneumonitis). The histologic finding of capillaritis is important to note, as it imparts a bad prognosis and is often treated more aggressively than simple steroid therapy.13 Because “acute lupus pneumonitis” is a clinical term, it is best avoided as a pathologic diagnosis. Instead, the type or types of lung injury should be noted, with special emphasis on the features of acute alveolar damage and capillaritis. Because the etiology of capillaritis is immune mediated, immunofluorescence for IgG and complement is sometimes a useful adjunct to diagnosis. A linear alveolar basement membrane deposit of IgG is suggestive of anti-GBM disease, and granular endothelial capillary deposition is suggestive of lupus-related capillaritis.
TABLE 50.3 Autoimmune Connective Tissue Diseases and Pulmonary Arterial Hypertension
Common (>20% of patients)
Systemic sclerosis
Mixed connective tissue disease
Not uncommon (>10% of patients)
Systemic lupus erythematosus
Uncommon (<10% of patients)
Dermatomyositis
Rheumatoid arthritis
Sjögren syndrome
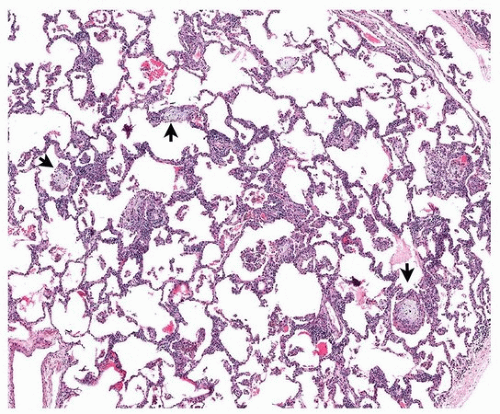
Chronic lupus pneumonitis is a clinical term for chronic interstitial lung disease in lupus patients and includes nonspecific interstitial pneumonia (the most common pattern) and usual interstitial pneumonia. Clinically, it is similar to idiopathic pulmonary fibrosis, with a better prognosis. Organizing pneumonia is a frequent histologic component, especially in combination with nonspecific interstitial pneumonia (Fig. 50.4).
FIGURE 50.1 ▲ Rheumatoid arthritis-associated interstitial lung disease, nonspecific interstitial pneumonia pattern. There is diffuse interstitial thickening with inflammation and fibrosis, without subpleural accentuation or spatial heterogeneity.
FIGURE 50.2 ▲ Rheumatoid nodule. There is an irregular area of necrotic debris mixed with fibrin, surrounded by acute and chronic inflammatory cells. The remainder of the specimen showed predominantly usual interstitial pneumonia pattern.
Chronic interstitial lung disease affects 3% to 8% of patients with systemic lupus, with a progressive increase in prevalence with disease duration, and is relatively less common than in other connective tissue disorders.13 In hospitalized patients with lupus, interstitial lung disease is seen in one-fourth of patients.15 In addition to usual and nonspecific interstitial pneumonia, lymphoid interstitial pneumonia has been described in occasional patients with lupus, usually in association with Sjögren syndrome. In these cases, the development of lung cysts by computed tomography is typical.13 Similar to acute lupus pneumonitis, an association between interstitial lung disease and the presence of anti-Ro/SSA autoantibodies has been observed for the chronic form of lung disease.15 In an autopsy study, the main pulmonary findings in patients dying with lupus included pleuritis and bacterial infections, including opportunistic infections, and pulmonary thromboembolism.16 Pulmonary hemorrhage occurred in one-fourth of patients, but fewer than 5% of the total showed capillaritis. An additional 5% of patient showed pulmonary hypertension with plexiform lesions.16
FIGURE 50.3 ▲ Capillaritis, systemic lupus erythematosus. There is patchy hemorrhage. The interstitium is markedly infiltrated by mixed inflammatory cells, including neutrophils.
FIGURE 50.4 ▲ Chronic interstitial inflammation with patchy organizing pneumonia, systemic lupus erythematosus. There is patchy interstitial inflammation and small foci of organizing pneumonia (arrows).
Pulmonary arterial hypertension develops more commonly in lupus patients than in individuals with rheumatoid arthritis, affecting 12% to 28% of patients. It is associated with Raynaud phenomenon, high levels of rheumatoid factor, and anti-RNP autoantibodies.13,17
Antiphospholipid Syndrome
The antiphospholipid syndrome is caused by circulating autoantibodies against phospholipid-binding plasma proteins that increase the risk of thrombosis and pregnancy loss. It is most frequently a primary syndrome but may be associated with other autoimmune diseases, most often systemic lupus erythematosus.
The pulmonary manifestations are presented in Table 50.4. The most common manifestation of lung disease in antiphospholipid syndrome is acute pulmonary embolism, which may often be the presenting symptom. Recurrent pulmonary emboli may give rise to pulmonary hypertension and chronic thromboembolic pulmonary hypertension. The prevalence of pulmonary hypertension in primary antiphospholipid syndrome and antiphospholipid syndrome associated with systemic lupus erythematosus has been estimated to be between 1.8% and 3.5%, respectively.18 Conversely, the prevalence of antiphospholipid autoantibodies in patients with chronic thromboembolic pulmonary hypertension varies between 10% and 20%.18
TABLE 50.4 Pulmonary Involvement in Antiphospholipid Syndrome (Patterns Often Coexist)
Clinical Diagnoses
Pathologic Diagnosis
Pulmonary embolism (acute or recurrent)
Pulmonary embolism
Pulmonary infarction
CTEPH
In situ thrombosis, acute, organizing, and recanalized