Left Ventricular Noncompaction Cardiomyopathy
Colin J. McMahon
Ricardo H. Pignatelli
Introduction
Left ventricular noncompaction cardiomyopathy (LVNC) is a genetic heterogeneous unique form of cardiomyopathy which has been reported on widely in the last few years (1,2,3,4,5,6). Some groups have reported it to comprise up to 9% of all pediatric cardiomyopathy (7). The existence, classification, and natural history of this form of cardiomyopathy remain controversial (8). This is added to by the lack of a diagnostic test and indeed discrepancies between groups in the classification of the disease. The increased reports of LVNC have resulted in speculation that the disease is in fact being overdiagnosed (9).
Definition
Left ventricular noncompaction is generally defined by (10):
Presence of prominent trabeculations in the left ventricle (Fig. 54.1 and Video 54.1).
Presence of deep recesses between the trabeculations and also presence of a thin compacted layer (Fig. 54.2 and Video 54.2).
Noncompacted: compacted ratio >2 in end systole on echocardiography.
The first two findings give a base to the actual imaging criteria for diagnosis of NCLV to be discussed later in this chapter.
History
Although Lewis first reported the presence of deep sinusoids in the left ventricle in 1904 (11), it was Grant who described the presence of multiple embryonal sinusoids in the left ventricular myocardium in 1926 (12,13). There are multiple other early reports of spongiform myocardium and abnormal myocardium with sinusoids, mostly based on pathology specimens (14,15,16,17,18,19,20,21). With subsequent improvements in imaging probe quality (i.e., better lateral resolution in ultrasound probes), and medical awareness of the issue, the diagnosis of NCLV has increased significantly. Adult studies have estimated its prevalence from 4 to 26 per 10,000 of the population (22,23). Pediatric studies have reported LVNC as the third most common cardiomyopathy following dilated and hypertrophic cardiomyopathy (5,24,25). The etiology of LVNC remains somewhat limited by the genetic heterogeneity, limited understanding of regulation of myocardial trabeculation, and compaction.
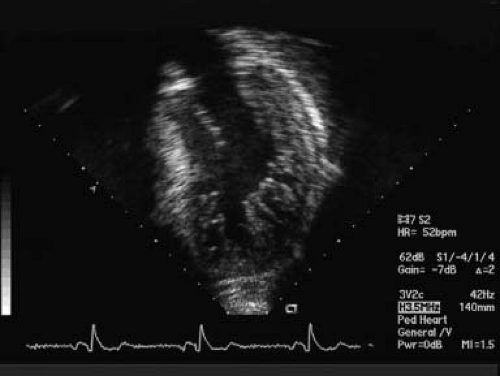 Figure 54.1 Classic echocardiographic image for diagnoses of NCLV. Apical view with presence of noncompacted segments in the lateral free wall of the LV and apex. NC/C ratio in systole: >2. |
Before reviewing normal early myocardial development one must recognize the increase of new and eye opening data challenging traditional concepts. To demonstrate this point are the continuous data concerning possible nonembryonic hypotheses as etiologies for NCLV. One of these hypotheses, that the increase of trabeculations may occur later in life, acquired NCLV, has been demonstrated in young athletes, forcing us to embrace this concept. Young competitors represent a clear example of having increased trabeculations arising as a result of myocardial remodeling (26) (Figs. 54.3 and 54.4; Video 54.3), yet the compacted layer is well developed. New de novo appearance of trabeculations which also is observed in pregnancy (27) could be an adaptive mechanism to increased loading conditions. Gati reported >25% de novo trabeculations during pregnancy (Fig. 54.5) (28). Finally in pediatric practice, we often encounter patients with sickle cell anemia presenting with an NCLV look-alike pattern on echocardiography (29). This may represent an accentuated
response by the myocardium to the increased cardiac preload (Fig. 54.6).
response by the myocardium to the increased cardiac preload (Fig. 54.6).
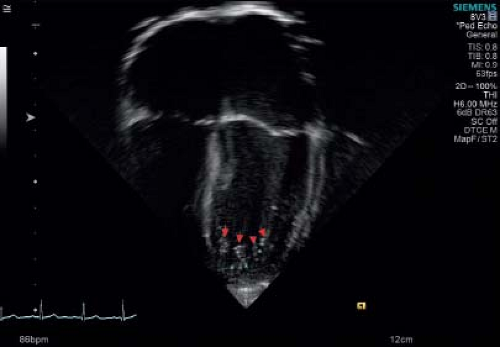 Figure 54.2 Red arrows depict tip of trabeculations. Green asterisks show deep recesses. |
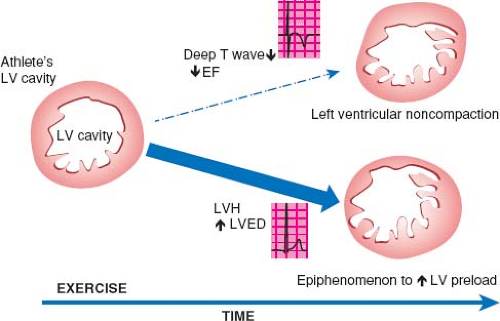 Figure 54.3 NCLV “acquired” in young athletes. (From Gati S, Chandra N, Bennett RL, et al. Increased left ventricular trabeculation in highly trained athletes: do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart. 2013;99:401–408.) |
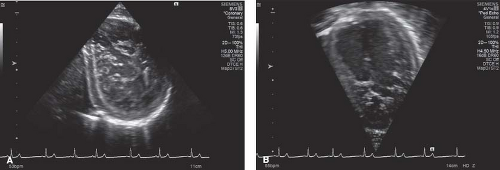 Figure 54.4 Asymptomatic African-American high school male whose only noteworthy information in medical history is the start (11 months) of weight lifting to qualify for the high school football team. A shows short axis and B shows its matching apical 4CH. |
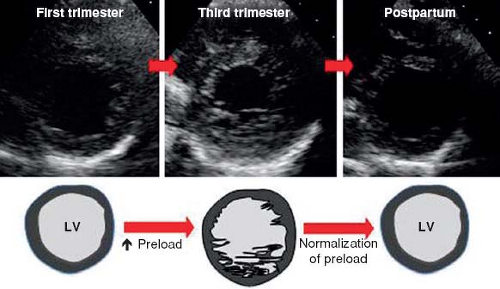 Figure 54.5 Diagram of proposed “de novo” trabeculations during pregnancy. (From Gati S, Papadakis M, Papamichael ND, et al. Reversible de novo left ventricular trabeculations in pregnant women: implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation. 2014;130:475–483.) |
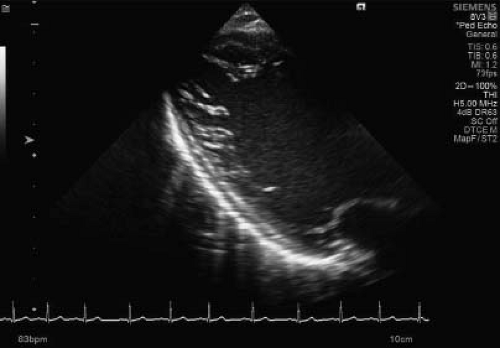 Figure 54.6 Long-axis view of a middle-aged young female with sickle cell disease. The long-axis view is highly suspicious for an increased number of trabeculations in the lateral wall of the LV. |
Recent valuable insights from Zhang et al. have highlighted some of the molecular mechanisms underlying normal and abnormal ventricular trabeculation and compaction (Fig. 54.7) (30).
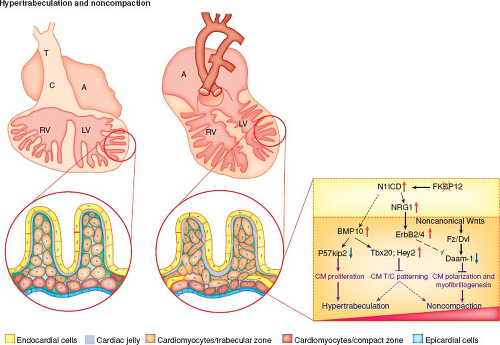 Figure 54.7 Hypertrabeculation and noncompaction. (From Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanisms of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet. 2013;163:144–156.) |
Normal Ventricular Myocardial Development
Ventricular myocardial trabeculation and subsequent compaction are required to generate functional myocardium. A specific sequence of morphogenetic steps occur in development of the normal ventricular myocardium (30). There are four stages in this development.
Formation of a single layer of ventricular myocardium. An early tubular heart is formed from the lateral mesoderm composed of three layers, one layer of myocardium, an endocardium, and a layer of cardiac jelly or extracellular matrix partitioning the two.
During the second stage, the myocardium thickens, endocardial cells invaginate, and myocardial cells along the inner wall form sheet protrusions into the lumen resulting in a trabecular myocardium. The outer layer of the myocardium will form the base for the compact myocardium. It has been proposed that the trabeculations allow nutrient and oxygen exchange within the myocardium and also augment heart muscle force generation in the developing embryo (31).
Myocardial compaction occurs at midgestation stage. The trabeculae compress toward the base of the wall of myocardium. This is completed by stage E14.5 in the mouse embryo.
Stage four is progression to a spiral multilayered mature myocardium during late fetal stage (32). Anderson et al. demonstrated that myocytes in the subendocardial and subepicardial layers
form helical sheets while the middle intervening layers form circular sheets (33). These characteristic myocyte arrangements are deemed important for the normal contractile process.
Multiple growth factors and signaling pathways in the endocardium are deemed important in this sequential process of hypertrabeculation, compaction, and maturation of the myocardium. P57kip2 is a cyclin-dependent kinase inhibitor of the p21 family, unique to the myocardium and its trabeculae and is believed to be crucial to suppression of trabeculation (34). Other growth factors include neuregulin 1 which is produced in endocardial cells and is mediated through myocardial receptors Erb2 and 4 (35). A study in zebrafish recently demonstrated that neuregulin 1 also regulates cardiac myocyte delamination to start trabeculation as well as promoting cell proliferation (36). Bone morphogenetic protein 10 (BMP10), vascular endothelial growth factor (VEGF), hyaluron synthase-2 (Has-2), and versican also regulate ventricular trabeculation and myocyte maturation (37,38). It has also become clear that the correct expression and amount of cardiac jelly, regulated by chromatin-remodeling factor Brg1 is required for normal development of the myocardium (39). Abnormal expression of Brg1 or ADAMTS1 may result in premature degeneration of cardiac jelly and cessation of trabeculation (40).
The epicardium also produces several mitogenic factors important in myocyte development. These include TGFB/BMPs, PDGF-A, Wnts, Hh, and FGFs and are important in development of the epicardium and the coronary circulation (41). Mice deficient in FGF9 or 16, or FGFR1 and 2 develop a thin myocardium (42,43). There is interaction between epicardial, myocardial, and endocardial signaling in the regulation of myocyte maturation. VCAM1 is specifically upregulated in cells exposed to epicardial growth factors while BMP10 and ANF are only upregulated by neuregulin 1 in the endocardium (44,45,46).
Ventricular Hypertrabeculation
FKBP12 is a cytoplasmic protein associated with BMP/activin/TGFB1 receptors, inositol triphosphate receptor, and the ryanodine receptor (47,48). Mice deficient in FKBP12 develop severe cardiac defects including increased trabecular number and thickness, recesses, lack of compaction and VSD; many features seen in LVNC (49). BMP10 is one of the genes upregulated in FKBP12-deficient hearts and its upregulation is associated with defective regulation of cardiac trabeculation and compaction (37). Pashmforoush et al. showed that upregulation of BMP10 in mouse hearts alone was sufficient to result in trabeculation abnormalities (38). Conversely BMP10-deficient mice died as a result of profoundly hypoplastic myocardial walls, deficient trabeculation, and cardiac failure (37). Cardiomyocyte proliferation is dramatically increased in FKBP12-deficient hearts and Nkx2.5 mutant hearts (37,38). P57kip2 is a negative cell cycle regulator and FKBP12-deficient mice express significantly lower levels of p57kip2 (34,50).
Watanabe et al. showed that overexpression of activated Notch1 in early cell lines leads to abnormal ventricular noncompaction (51). Notch1-activated protein is found in the endocardial cells at the base of trabeculae (52). BMP10 is required for Notch1-mediated myocyte regulation (53). BMP10 also regulates Tbx20 myocardial expression (54). Thus notch signaling appears to be a critical factor in normal trabecular/compaction development.
Other signaling components including Wnt/PCP mediate normal trabeculation and compaction via Vangl2 and Scrib molecules (55,56). Normal myocyte polarization is important in development of the myocardium. Daam-1, also downregulated in FKBP12-deficient mice is crucial for normal myocyte patterning (57).
Two existing possible embryologic morphogenesis hypotheses have been proposed by Arbustini (26).
One is an arrest or abnormal myocardial morphogenesis causing NCLV, occurring during heart development when myocyte development fails to evolve from spongiform stage to the compacted stage.
Cayre speculated that NCLV is not due to an arrest of myocardial compaction only, but an alteration of this process, starting a morphogenetic spectrum which drives from a delay to an excess of the compaction process and would determine the different evolutionary types of this disease (58).
Arbustini’s second theory proposes that NCLV occurs due to inhibition of the regression of embryonic structures resulting in a looseness of cells or cell bundles giving rise to spongiform myocardium (26).
Genetic Causes of LVNC
Several genes have been implicated in the development of LVNC (Table 54.1) (59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79). These primarily involve the sarcomere proteins, typically associated with cardiac contractility, rather than crucial stages of embryologic development between trabeculation and compaction. Phoon et al. studied the role of tafazzin (G4.5) in the development of cardiomyopathy (68). Tafazzin knockout mice developed altered cardiolipin and mitochondrial microstructure and the hearts had an LVNC type phenotype. This provides some evidence for LVNC in Barth syndrome.
Similarly, mutations in troponin T gene (TNNT2) have been reported in a familial case of LVNC (78). The potential for TNNT2 gene mutations to result in differing cardiomyopathic phenotypes prompted the suggestion of genetic modifiers in the pathogenesis of LVNC. One such modifier Mib1, in knockout mice resulted in an LVNC phenotype (79). Interestingly Mib1, regulates Notch1 ligands Jagged and Delta, once again highlighting the role of notch signaling in regulation of normal ventricular myocardial wall development. Recently HCN4 mutations have been identified in patients with LVNC and bradycardia (74,75). Hoedemaekers et al. reported LVNC to be a genetic condition when combining cardiologic family testing and molecular testing in 67% of patients (80). Probst et al. reported sarcomere gene mutations present in 29% of cases of isolated LVNC but there was no correlation between genetic mutation and clinical phenotype (81).
Chromosomal Conditions Associated with LVNC
In addition to several gene mutations associated with LVNC, several chromosomal disorders and clinical diagnoses have been described in this cardiomyopathy (82,83,84,85,86,87,88,89,90,91,92,93,94).
TABLE 54.1 Genes Involved in LVNC | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Clinical Presentation
LVNC may be asymptomatic or can present with heart failure, arrhythmias (especially ventricular), and/or thromboembolic events (95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123). Familial disease occurs in 18% to 50% of adults with isolated LVNC (7,124,125). There is often intrafamilial phenotypic variability including HCM, DCM, and LVNC further promoting the concept of a final common pathway for cardiomyopathies (126,127,128).
Brescia et al. reviewed 242 pediatric patients with isolated LVNC and reported ECG abnormalities, most commonly hypertrophy and repolarization, in 87% of cases (129). Death occurred in 12.8%; 5.4% underwent transplantation. Sudden cardiac death occurred in 6.2%, most commonly associated with ventricular arrhythmias and cardiac dysfunction. There were no deaths in patients with normal cardiac dimensions, function, and normal cardiac rhythm.
Undulating Phenotype
LVNC may alter phenotype over time changing from a normal phenotype to either a dilated or hypertrophic phenotype (7). Likewise, ventricular systolic function may initially deteriorate then improve, only to subsequently deteriorate later in life again (7). This undulating phenotype and function appears to be unique to LVNC. Saleeb et al. observed the same undulating phenotype in a cohort of 52 patients (Fig. 54.8) (130).
Prenatal Diagnosis
Several authors have reported on the antenatal diagnosis of LVNC (131,132,133,134,135). Diagnosis is most commonly made from the apical four-chamber view. There may be variable degrees of ventricular dysfunction including well-preserved systolic function in antenatally diagnosed cases. LVNC may occur in isolation or in conjunction with congenital cardiac defects (135).
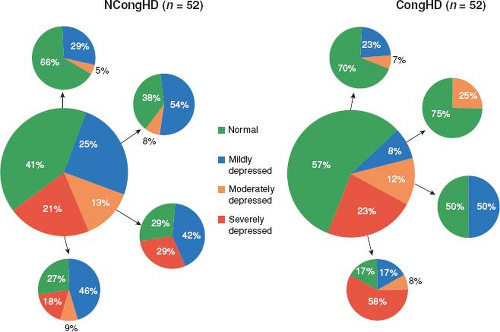 Figure 54.8 Changes of LV function since first echocardiogram. (From Saleeb SF, Margossian R, Spencer CT, et al. Reproducibility of echocardiographic diagnosis of left ventricular noncompaction. J Am Soc Echocardiogr. 2012;25:194–202.) |
LVNC with Normal Ejection Fraction
Several patients with clear evidence of hypertrabeculation may have normal systolic and diastolic function (136). There is some overlap with children who have more than three trabeculations but normal phenotype and normal left ventricular systolic and diastolic function. One needs to be careful not to overcall LVNC in this setting as there may be some patients with LV trabeculation who are in fact normal. Echocardiographic assessment of ejection fraction using Simpson method in this population may be challenging given the extensive degree of trabeculations (137). Consistency in measurement of left ventricular ejection fraction (LVEF) may also be challenging depending on criteria for including or excluding trabeculations.
Noncardiac Manifestations of LVNC
Several investigators have emphasized the importance of assessing extracardiac manifestations in patients with LVNC (138,139,140). Stöllberger et al. have promoted neurologic evaluation and assessment for skeletal myopathy (141). Other organ systems that are involved include thyroid and auditory abnormalities. Similarly Pignatelli et al. and Scaglia et al. highlighted the association with mitochondrial disorders in several children with LVNC and advocated skeletal muscle biopsy in such patients (7,142). We would advocate that brain MRI and skeletal muscle biopsy be considered in cases suspicious for underlying mitochondrial abnormalities and developmental delay.
There is a high prevalence of LVNC in Duchenne and other forms of muscular dystrophy often associated with progressive
reduction in LV systolic function over time (Fig. 54.9; Videos 54.4 and 54.5) (138,143,144,145,146,147).
reduction in LV systolic function over time (Fig. 54.9; Videos 54.4 and 54.5) (138,143,144,145,146,147).
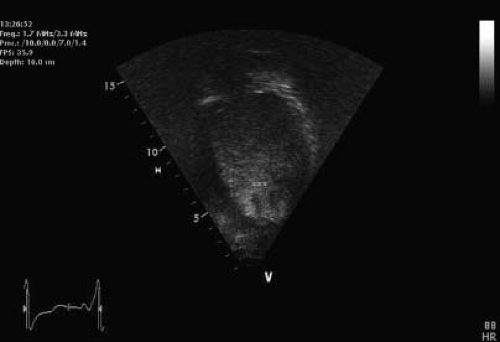 Figure 54.9 A 10-year-old boy with a diagnosis of Duchenne, NCLV, and severe PHT. Contrast negative to rule out apical thrombosis. Several trabeculations are appreciated (*). |
Electrocardiogram Findings
ECG findings often include massive biventricular hypertrophy which should be distinguished from Pompe disease (7). Other electrocardiographic findings include first-, second-, and third-degree atrioventricular block and bradycardia; the latter associated with HCN4 mutations (76,77,148,149,150,151). Supraventricular and ventricular tachycardia are also reported findings on Holter or telemetry monitoring (152,153,154,155,156). An alternating LBBB and RBBB have been described in association with septal ventricular tachycardia (157). There are reports of placement of a cardioverter-defibrillator for ventricular fibrillation in the setting of LVNC (158,159).
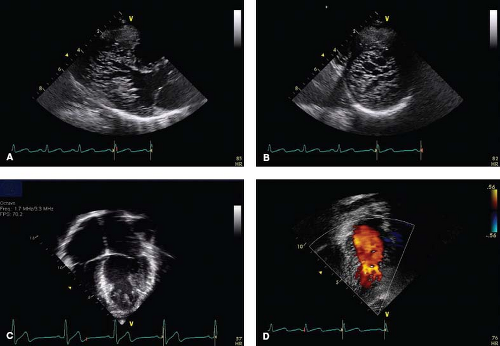 Figure 54.10 A series of echo images and projections highly suggestive of NCLV. |
Echocardiographic Assessment of LVNC
Echocardiography is the first tool used for establishing the diagnosis of LVNC (7,10,159,160,161,162,163,164,165,166). Standard 2-D imaging demonstrates trabeculations and deep intertrabecular recesses or “valleys” in the left ventricle (Fig. 54.10 and Videos 54.6 to 54.9) (7,10).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


