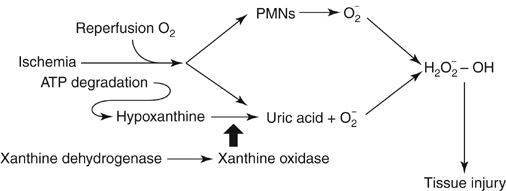
Regardless of the etiology of the limb ischemia—embolus, thrombosis, or trauma—skeletal muscle ischemia is the inciting event (Box 1). This results in deletion of cellular energy stores within the skeletal muscle cell, leading to impaired cellular ion homeostasis and increased cell membrane permeability. From this ensues an anaerobic process that can ultimately result in cellular dysfunction and death. However, the extent of the injury is not just limited by the ischemic time period. Key to understanding of the concept of ischemia–reperfusion injury is the paradoxical increase in cellular injury associated with reinstitution of blood flow into ischemic tissues. With reperfusion and reintroduction of oxygen, oxygen free radicals form, exacerbating tissue injury and further increasing capillary permeability. Tissue hypoxia also results in the mobilization of neutrophils into the interstitium with potential for deleterious effects upon the endothelial barrier. The increased generation of reactive oxygen metabolites (activated oxygen free radicals) during reperfusion compromises the production of nitric oxide and prostaglandins, favoring vasoconstriction and further limiting overall perfusion. The source of activated oxygen free radicals may be exogenous, from activated neutrophils, or may be produced endogenously, from within the muscle tissue. For skeletal muscle ischemia, the primary endogenous source of oxygen free radicals is the xanthine dehydrogenase–xanthine oxidase pathway. During ischemia, calcium-activated proteases convert the enzyme xanthine dehydrogenase to xanthine oxidase (Figure 1). In the presence of oxygen, now reestablished through reperfusion, hypoxanthine, a byproduct of adenosine triphosphate degradation, is converted by xanthine oxidase to uric acid and the oxygen free radical superoxide. These generated free radicals cause injury through chain reaction lipid peroxidation and inactivation of various cellular enzymes, destabilizing cell membranes.
Ischemia-Induced Muscle Myonecrosis, Myoglobinuria, and Secondary Kidney Failure
Pathophysiology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine
