Interstitial Lung Disease
GENERAL PRINCIPLES
Definition
• Interstitial lung disease (ILD) describes a heterogeneous group of over 200 diseases affecting the pulmonary interstitium with varying degrees of involvement of the pleural space, airways, and pulmonary vasculature.
• ILD is also termed diffuse parenchymal lung disease (DPLD).
• These diseases account for ∼15–20% of general pulmonary practice.
• Since ILDs differ greatly in presentation, clinical course, and response to therapy, establishing an accurate diagnosis is essential for determining the optimal management strategy.
• This requires effective collaboration between the pulmonologist, thoracic surgeon, radiologist, and pathologist to integrate clinical, physiologic, laboratory, radiographic, and histopathologic data.
• Accordingly, the majority of this chapter will focus on the clinical evaluation and diagnosis of ILD.
Classification
• Guidelines based on clinical, histopathologic, and radiographic findings have been proposed for subgroups of ILD including idiopathic interstitial pneumonias (IIP), hypersensitivity pneumonitis (HP), lymphangioleiomyomatosis (LAM), and others.
• However, due to the heterogeneous nature of these diseases, there is currently no universal classification system that encompasses all ILDs.
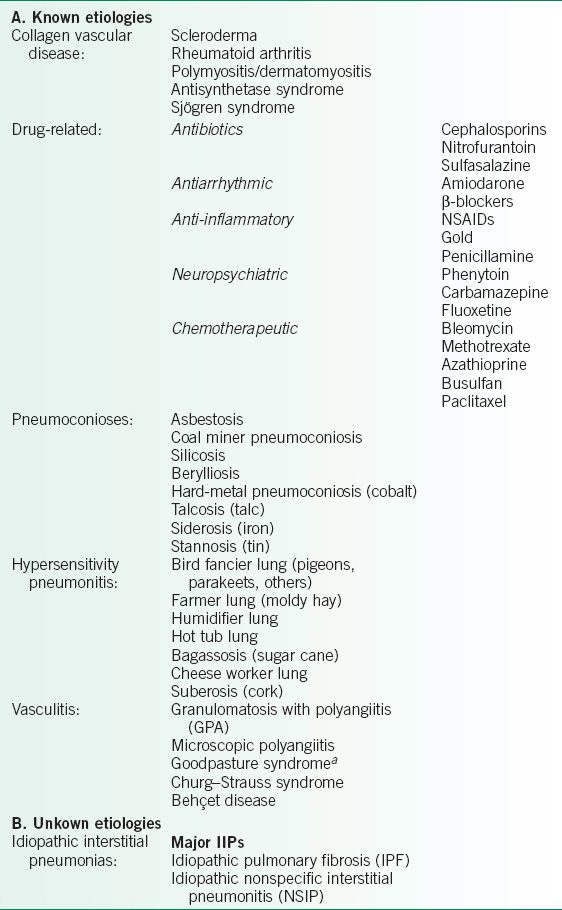
• A classification system based loosely on etiology and/or disease association is presented in Table 25-1.
• Keep in mind that this table is far from comprehensive, and the definition and classification of ILDs will continue to evolve rapidly as we learn more about the pathogenesis of these diseases.
Etiology and Pathogenesis
• Environmental and heritable factors both play a significant role in the pathogenesis of ILD, but the relative contribution and importance of these factors is quite variable between diseases and patients.
• An increasing number of occupational/environmental exposures and genetic modifiers of disease susceptibility have been elucidated through epidemiologic and genomic analyses, respectively.
• However, the complex interactions between these factors and how they affect disease development and progression remain poorly understood.
• Alveolar epithelial cell injury is a hallmark of ILD. The source of injury may be extrinsic, as in cases of HP, pneumoconiosis, or radiation pneumonitis.
• Alternatively, the injurious insult may arrive via the circulation, as suspected in collagen vascular diseases, vasculitides, or drug-induced lung diseases.
• Normally after a limited injury, the initial acute inflammatory response resolves, and tissue repair programs restore lung integrity and homeostasis.
• However, with recurrent or persistent injury the reparative response becomes maladaptive; this leads to dysregulation of the normal injury repair response, resulting disruption of lung architecture and function.
• Indeed, lung biopsy specimens from patients with ILD frequently show varying degrees of inflammation and/or fibrosis.
• In addition to recurrent injury and aberrant repair of the airway epithelium, other factors may contribute to the pathogenesis of ILD.
• Studies have revealed an association between short telomere length and idiopathic pulmonary fibrosis (IPF); these findings are consistent with the increased prevalence of IPF in elderly patients and implicate accelerated cellular aging or stem cell exhaustion as additional mechanisms of disease in some ILDs.
TABLE 25-1 CLASSIFICATION OF DIFFUSE PROLIFERATIVE LUNG DISEASES

DIAGNOSIS
• A suggested algorithm for the evaluation of ILD is presented in Figure 25-1.
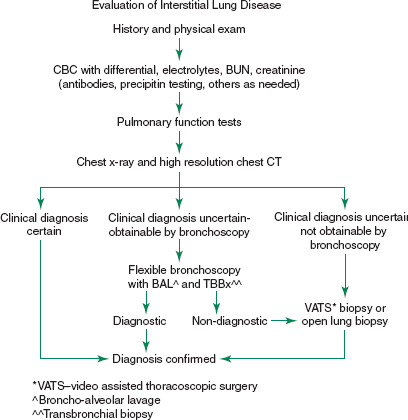
FIGURE 25-1. Evaluation of diffuse proliferative lung diseases. (Adapted from American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002;165:277–304; and British Thoracic Society and Standards of Care Committee. The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. Thorax. 1999;54(Suppl 1):S1–S28.)
Clinical Presentation
History
• A comprehensive history is a very important part of the patient evaluation.
• It can significantly narrow the differential diagnosis, guide the physical examination, and reduce the need for extensive diagnostic testing.
• Dyspnea is the most common presenting symptom.
 Patients may initially present with dyspnea only with moderate or heavy exertion.
Patients may initially present with dyspnea only with moderate or heavy exertion.
 As the disease progresses, breathlessness with mild or minimal exertion becomes apparent.
As the disease progresses, breathlessness with mild or minimal exertion becomes apparent.
 Eventually patients become dyspneic at rest.
Eventually patients become dyspneic at rest.
 Depending on the specific disease, dyspnea may present insidiously over months to years as in IPF, or pursue a more aggressive course over weeks to months as in acute interstitial pneumonia or acute eosinophilic pneumonia.
Depending on the specific disease, dyspnea may present insidiously over months to years as in IPF, or pursue a more aggressive course over weeks to months as in acute interstitial pneumonia or acute eosinophilic pneumonia.
 Episodic dyspnea may occur in cases of HP where repeated exposure to an inciting environmental agent causes waxing and waning symptoms.
Episodic dyspnea may occur in cases of HP where repeated exposure to an inciting environmental agent causes waxing and waning symptoms.
 Therefore, it is important to accurately quantify the duration and severity of the patient’s dyspnea.
Therefore, it is important to accurately quantify the duration and severity of the patient’s dyspnea.
• Cough is also a frequent complaint associated with ILD.
 A nonproductive cough is common in cases of IPF, HP, and sarcoidosis.
A nonproductive cough is common in cases of IPF, HP, and sarcoidosis.
• Chest pain is unusual.
 When present it may be associated with inflammation of the pleural space (systemic lupus erythematosus, rheumatoid arthritis), pneumothorax (LAM), or an atypical cause of ILD (sarcoidosis).
When present it may be associated with inflammation of the pleural space (systemic lupus erythematosus, rheumatoid arthritis), pneumothorax (LAM), or an atypical cause of ILD (sarcoidosis).
 Occult coronary artery disease is common in elderly patients with advanced lung disease and limited functional capacity. Therefore, coronary ischemia should be considered in the differential.
Occult coronary artery disease is common in elderly patients with advanced lung disease and limited functional capacity. Therefore, coronary ischemia should be considered in the differential.
• Wheezing is less frequent in general.
 It may be more prevalent in ILDs involving the airways such as HP, respiratory bronchiolitis-interstitial lung disease (RB-ILD), or sarcoidosis.
It may be more prevalent in ILDs involving the airways such as HP, respiratory bronchiolitis-interstitial lung disease (RB-ILD), or sarcoidosis.
 Other diseases involving the airways, such as chronic bronchitis or asthma, may occur concurrently.
Other diseases involving the airways, such as chronic bronchitis or asthma, may occur concurrently.
• Hemoptysis is also infrequent. It may occur in ILDs associated with vasculitis, connective tissue diseases or diffuse alveolar hemorrhage, such as Goodpasture syndrome, microscopic polyangiitis, and granulomatosis with polyangiitis (GPA, known previously as Wegener granulomatosis).
• Constitutional symptoms, such as fevers, chills, weight loss, night sweats, and fatigue occur with variable frequency. Significant unintentional weight loss should also raise the possibility of a concurrent malignancy, since patients with certain ILDs such as IPF, lymphoid interstitial pneumonia (LIP), and asbestosis are known to have an increased incidence of lung malignancy.
• Past and current medical histories are important for diagnosing ILDs secondary to systemic conditions such as collagen vascular disease, vasculitides, and other autoimmune diseases (Table 25-1).
 In some cases, the systemic disease is already present at the time of diagnosis.
In some cases, the systemic disease is already present at the time of diagnosis.
 However, in certain ILDs associated with systemic disease, ILD may be the initial manifestation of the disease.
However, in certain ILDs associated with systemic disease, ILD may be the initial manifestation of the disease.
 In rare cases ILD can be the only manifestation of the disease.
In rare cases ILD can be the only manifestation of the disease.
 The clinician should maintain a high index of clinical suspicion, and in the correct clinical context, such as a younger female presenting with ILD, diagnostic testing should be performed to exclude the presence of collagen vascular disease.
The clinician should maintain a high index of clinical suspicion, and in the correct clinical context, such as a younger female presenting with ILD, diagnostic testing should be performed to exclude the presence of collagen vascular disease.
• Social history should be obtained to identify known risk factors for certain ILDs.
 Cigarette smoking has an integral causal relationship with diseases such as RB-ILD, desquamative interstitial pneumonia (DIP), and pulmonary Langerhans cell histiocytosis (PLCH).
Cigarette smoking has an integral causal relationship with diseases such as RB-ILD, desquamative interstitial pneumonia (DIP), and pulmonary Langerhans cell histiocytosis (PLCH).
 In addition, diffuse alveolar hemorrhage occurs in nearly 100% of patients with Goodpasture syndrome who smoke and only around 20% of those who do not.
In addition, diffuse alveolar hemorrhage occurs in nearly 100% of patients with Goodpasture syndrome who smoke and only around 20% of those who do not.
 Conversely, ILDs such as HP, chronic eosinophilic pneumonia, and sarcoidosis appear to be less common in cigarette smokers.
Conversely, ILDs such as HP, chronic eosinophilic pneumonia, and sarcoidosis appear to be less common in cigarette smokers.
 Recreational drug abuse has also been described as a cause of ILD.
Recreational drug abuse has also been described as a cause of ILD.
• Occupational and environmental histories are an essential part of any DLPD workup.
 A large number of occupational and environmental exposures have been implicated as causative agents for ILDs.
A large number of occupational and environmental exposures have been implicated as causative agents for ILDs.
 The occupational history should cover the patient’s entire lifetime since the time between exposure and disease onset may span many years.
The occupational history should cover the patient’s entire lifetime since the time between exposure and disease onset may span many years.
 Details of each exposure, including duration, frequency, intensity, and presence or absence of respiratory protection should be recorded.
Details of each exposure, including duration, frequency, intensity, and presence or absence of respiratory protection should be recorded.
 The occupational history of close contacts should also be established, as exposure to an inciting agent (e.g., asbestos) may not occur only in the workplace.
The occupational history of close contacts should also be established, as exposure to an inciting agent (e.g., asbestos) may not occur only in the workplace.
 Environmental (nonwork-related) exposures including pets, hobbies, and recreational activities should also be reviewed.
Environmental (nonwork-related) exposures including pets, hobbies, and recreational activities should also be reviewed.
 Finally, a detailed travel and residential history should also be obtained.
Finally, a detailed travel and residential history should also be obtained.
• Family history may be important in certain ILDs with a known heritable basis. These include diseases like familial IPF, Hermansky–Pudlak syndrome (HPS), and lysosomal storage disorders such as Gaucher disease and Niemann–Pick disease.
• Therapeutic agents are a common cause of ILD.
 Therapeutic agents include not only prescription medications, but also over-the-counter medications, herbal supplements, radiation therapy for malignant diseases, and other forms of therapy the patient may have been receiving.
Therapeutic agents include not only prescription medications, but also over-the-counter medications, herbal supplements, radiation therapy for malignant diseases, and other forms of therapy the patient may have been receiving.
 Development of disease may occur years after the initial exposure.
Development of disease may occur years after the initial exposure.
 Thus it is important to obtain not only a list of current medications but also a comprehensive therapeutic agent history.
Thus it is important to obtain not only a list of current medications but also a comprehensive therapeutic agent history.
Physical Examination
• Because the pulmonary examination in patients with ILD is nonspecific, the main goal of the physical examination is to search for evidence of systemic disease that may help narrow the differential diagnosis.
• Extrathoracic manifestations of systemic diseases such as collagen vascular disease, amyloidosis, sarcoidosis, and vasculitis may be present.
• Head and neck examination should exclude:
 enlarged lachrymal, parotid, and salivary glands (sarcoidosis and systemic sclerosis)
enlarged lachrymal, parotid, and salivary glands (sarcoidosis and systemic sclerosis)
 conjunctivitis and episcleritis (collagen vascular diseases and sarcoidosis)
conjunctivitis and episcleritis (collagen vascular diseases and sarcoidosis)
 dry mouth or eyes (primary or secondary Sjögren syndrome)
dry mouth or eyes (primary or secondary Sjögren syndrome)
 lymphadenopathy (sarcoidosis, lymphoma)
lymphadenopathy (sarcoidosis, lymphoma)
 lupus pernio (sarcoidosis)
lupus pernio (sarcoidosis)
 alopecia (systemic lupus erythematosus, sarcoidosis)
alopecia (systemic lupus erythematosus, sarcoidosis)
• Pulmonary examination is most commonly characterized by bilateral fine inspiratory crackles often described as Velcro crackles. Other findings on lung examination may include coarse crackles, and less commonly, wheezing.
• Cardiovascular examination should focus on detecting signs of pulmonary hypertension and right ventricular dysfunction, including:
 elevated jugular venous pressure and peripheral pitting edema
elevated jugular venous pressure and peripheral pitting edema
 pulsatile liver and hepatojugular reflux
pulsatile liver and hepatojugular reflux
 right ventricular heave, accentuated second heart sound (P2), and tricuspid regurgitation
right ventricular heave, accentuated second heart sound (P2), and tricuspid regurgitation
 Left ventricular dysfunction or valvular dysfunction may also be present in systemic diseases such as amyloidosis, sarcoidosis, and Behçet disease
Left ventricular dysfunction or valvular dysfunction may also be present in systemic diseases such as amyloidosis, sarcoidosis, and Behçet disease
• Abdominal examination may reveal hepatomegaly and/or splenomegaly in collagen vascular diseases, amyloidosis, sarcoidosis, or lymphoma.
• Musculoskeletal examination may reveal digital clubbing, arthritis, effusions, joint deformities, contractures, muscle atrophy, swelling, tenderness, or weakness.
• Skin examination may show nonpitting edema, sclerosis, various rashes, purpura, SC nodules, telangiectasias, calcinosis, abnormal pigmentation, plaques, or ulcerations from digital ischemia.
• Neurologic examination may reveal a broad spectrum of central and peripheral deficits ranging from subtle cognitive defects to peripheral neuropathy and paresthesias, mononeuritis multiplex, autonomic dysfunction, or focal deficits associated with cerebral ischemia.
Diagnostic Testing
Laboratory Testing
• Laboratory testing for ILDs should be directed by history and physical examination findings.
• General testing should include:
 complete blood count with differential
complete blood count with differential
 renal function panel
renal function panel
 hepatic function panel
hepatic function panel
 urinalysis (where indicated)
urinalysis (where indicated)
• Testing for collagen vascular diseases should be conducted in the appropriate clinical context. Where indicated, the following labs should be drawn:
 Antinuclear antibodies (ANA) and extractable nuclear antigens (ENA)
Antinuclear antibodies (ANA) and extractable nuclear antigens (ENA)
 Rheumatoid factor (RF) and anticyclic citrullinated peptide antibodies (anti-CCP3)
Rheumatoid factor (RF) and anticyclic citrullinated peptide antibodies (anti-CCP3)
 Creatine kinase (CK), aldolase, and anti-Jo1 antibodies
Creatine kinase (CK), aldolase, and anti-Jo1 antibodies
 Scl-70 and anticentromere antibodies
Scl-70 and anticentromere antibodies
 Double-stranded DNA antibodies
Double-stranded DNA antibodies
 Myositis panel
Myositis panel
• Routine use of serum angiotensin-converting enzyme (ACE) levels in sarcoidosis is not recommended as ACE levels have a poor sensitivity and specificity for diagnosing the disease. Moreover, ACE levels correlate poorly with radiographic findings and physiologic impairment, and have no prognostic utility.
• Serum precipitin testing in HP may be used to confirm the presence of serum antibodies against a specific antigen implicated as a causative agent of disease.
 However, the presence of specific circulating antibodies only serves as evidence of exposure, and does not confirm that the agent is responsible for the disease.
However, the presence of specific circulating antibodies only serves as evidence of exposure, and does not confirm that the agent is responsible for the disease.
 Furthermore, antigen panels can differ significantly between institutions and are usually directed at antigens commonly seen in each particular community. As a result, these tests often do not identify novel or rare antigens.
Furthermore, antigen panels can differ significantly between institutions and are usually directed at antigens commonly seen in each particular community. As a result, these tests often do not identify novel or rare antigens.
Physiologic Testing
• Pulmonary function tests (PFTs) in ILDs such as IPF are classically described as having a purely restrictive pattern (see Chapter 3).
 However, the finding of restriction on PFTs is nonspecific and may be due to a number of causes including chest wall disease, obesity, neuromuscular disease, etc.
However, the finding of restriction on PFTs is nonspecific and may be due to a number of causes including chest wall disease, obesity, neuromuscular disease, etc.
 In reality, a significant number of ILDs show a mixed obstructive–restrictive pattern on pulmonary function testing.
In reality, a significant number of ILDs show a mixed obstructive–restrictive pattern on pulmonary function testing.
 A predominantly obstructive pattern may be seen in ILDs with small airways involvement including sarcoidosis, HP, and the smoking-related ILDs like PLCH, RB-ILD, and DIP.
A predominantly obstructive pattern may be seen in ILDs with small airways involvement including sarcoidosis, HP, and the smoking-related ILDs like PLCH, RB-ILD, and DIP.
 Combined pulmonary fibrosis and emphysema (CPFE) may present with normal appearing PFTs, however, the diffusion capacity (DLCO) is universally decreased in these patients.
Combined pulmonary fibrosis and emphysema (CPFE) may present with normal appearing PFTs, however, the diffusion capacity (DLCO) is universally decreased in these patients.
• DLCO in patients with ILDs is almost invariably reduced. This reduction may be due to a number of factors depending on the etiology of the ILD, including abnormal V/Q relationships, decreased surface area for gaseous diffusion, and in extreme cases, a thickened alveolar–capillary interface.
• The 6-minute walk test is a useful tool in the evaluation of ILD.
 It provides a measurement of a patient’s exercise capacity, and can be used to follow a patient’s disease progression and/or response to therapy.
It provides a measurement of a patient’s exercise capacity, and can be used to follow a patient’s disease progression and/or response to therapy.
 Moreover, it has prognostic value in diseases like IPF.
Moreover, it has prognostic value in diseases like IPF.
 A decrease of ≥5% points (e.g., from 95% to 90%) with exertion is generally considered to be a significant physiologic drop in oxygen saturation.
A decrease of ≥5% points (e.g., from 95% to 90%) with exertion is generally considered to be a significant physiologic drop in oxygen saturation.
• Oxygen assessments are based on 6-minute walking tests. They allow for assessment of a patient’s supplemental oxygen requirements at rest and with exertion.
Imaging
Plain Film CXR
• Despite the advent of high-resolution computed tomography (HRCT), plain film CXR is still a useful modality for evaluating DLPD.
• It is not uncommon for findings on CXR to predate the clinical presentation, sometimes by 5–10 years.
• In some cases, subclinical ILD may be incidentally detected on a CXR obtained for unrelated reasons in an otherwise asymptomatic patient.
• Thus, reviewing old studies as part of the initial evaluation may yield useful information on the disease course and progression.
• Certain ILDs have characteristic appearance on CXR that can assist significantly in narrowing the differential diagnosis, including:
 LAM
LAM
 PLCH
PLCH
 silicosis
silicosis
 asbestosis
asbestosis
 sarcoidosis
sarcoidosis
 chronic eosinophilic pneumonia
chronic eosinophilic pneumonia
• Markings commonly found in ILD include linear (reticular) markings, nodules, opacities, and honeycombing.
 Honeycombing suggests an end-stage fibrotic process, which may be the result of progression of any number of diseases (IPF, HP, sarcoidosis, and scleroderma).
Honeycombing suggests an end-stage fibrotic process, which may be the result of progression of any number of diseases (IPF, HP, sarcoidosis, and scleroderma).
 Ground-glass opacity, an increased attenuation of lung parenchyma that does not obscure pulmonary vessels, can be found in both interstitial (nonspecific interstitial pneumonitis [NSIP], DIP, sarcoidosis) and alveolar diseases.
Ground-glass opacity, an increased attenuation of lung parenchyma that does not obscure pulmonary vessels, can be found in both interstitial (nonspecific interstitial pneumonitis [NSIP], DIP, sarcoidosis) and alveolar diseases.
 Linear (or reticular) markings (sarcoidosis, pneumoconiosis, NSIP, IPF) are almost always associated with interstitial processes.
Linear (or reticular) markings (sarcoidosis, pneumoconiosis, NSIP, IPF) are almost always associated with interstitial processes.
 Nodular markings (pneumoconiosis, sarcoidosis, PLCH, GPA) also have a strong association with interstitial processes.
Nodular markings (pneumoconiosis, sarcoidosis, PLCH, GPA) also have a strong association with interstitial processes.
• The presence of pleural disease on CXR may also be helpful since pleural involvement in ILD is generally uncommon.
• Diseases that affect the pleura include:
 collagen vascular diseases (pleural effusion, pleural thickening)
collagen vascular diseases (pleural effusion, pleural thickening)
 asbestosis (pleural plaques, pleural calcifications, mesothelioma)
asbestosis (pleural plaques, pleural calcifications, mesothelioma)
 LAM (pneumothorax and chylous effusions)
LAM (pneumothorax and chylous effusions)
 PLCH (pneumothorax)
PLCH (pneumothorax)
• The distribution of interstitial markings on CXR can be useful because certain diseases have a predilection for affecting particular areas of the lung.
• In general, diseases can be grouped into those affecting predominantly the upper lobes and those affecting predominantly the lower.
 Diseases affecting the upper lobes include CF, ankylosing spondylitis, sarcoidosis, silicosis, eosinophilic granuloma (PLCH), TB, Pneumocystis jiroveci pneumonia, Crohn disease-associated ILD, ulcerative colitis-associated ILD, and ILD secondary to bischloroethylnitrosourea (BCNU) chemotherapy.
Diseases affecting the upper lobes include CF, ankylosing spondylitis, sarcoidosis, silicosis, eosinophilic granuloma (PLCH), TB, Pneumocystis jiroveci pneumonia, Crohn disease-associated ILD, ulcerative colitis-associated ILD, and ILD secondary to bischloroethylnitrosourea (BCNU) chemotherapy.
 These can be remembered with the mnemonic CASSET-P-CUB.
These can be remembered with the mnemonic CASSET-P-CUB.
 Diseases affecting the lower lobes include bronchiectasis, asbestosis, lymphangitic carcinomatosis, DIP/usual interstitial pneumonia (UIP)/NSIP, aspiration, sarcoidosis (note, also included under upper lobe diseases), and scleroderma-associated ILD.
Diseases affecting the lower lobes include bronchiectasis, asbestosis, lymphangitic carcinomatosis, DIP/usual interstitial pneumonia (UIP)/NSIP, aspiration, sarcoidosis (note, also included under upper lobe diseases), and scleroderma-associated ILD.
 These can be remembered with the mnemonic BALDASS.
These can be remembered with the mnemonic BALDASS.
• Despite the aforementioned advantages of CXR in evaluating ILD, it is also important to remember that almost 10% of patients with biopsy-proven diffuse lung disease may have a normal CXR.
• Also, the clinical severity of ILDs may be difficult to predict from radiographic findings.
• In IPF, for example, the clinical severity of disease is often greater than would be predicted by the CXR.
• The converse is often true in the case of nodular diseases such as sarcoidosis, LCH, and pneumoconiosis, in which patients can be asymptomatic despite radiographic abnormalities.
High-Resolution Computed Tomography
• HRCT has revolutionized the evaluation of ILDs, as it offers far greater spatial resolution than CXR.
• The diagnostic power of HRCT scanning has increased substantially as a result of clinical, histopathologic, and radiographic experience accumulated over the last 20 years and advances in scanner technology.
• As such, characteristic HRCT findings have become diagnostic in certain ILDs, enabling clinicians to make a diagnosis with a high degree of confidence without the need for surgical lung biopsy (SLB).
• One such disease is IPF.
 IPF is a lethal fibrosing interstitial pneumonia of unknown etiology characterized by progressive pulmonary fibrosis associated with histopathologic pattern of UIP.
IPF is a lethal fibrosing interstitial pneumonia of unknown etiology characterized by progressive pulmonary fibrosis associated with histopathologic pattern of UIP.
 Histopathologic UIP pattern is defined as patchy involvement of the lung parenchyma by fibrosis with architectural distortion, honeycombing in a predominantly subpleural/paraseptal distribution, presence of fibroblastic foci, and absence of features suggesting an alternative diagnosis such as granulomas, organizing pneumonia, or hyaline membranes.
Histopathologic UIP pattern is defined as patchy involvement of the lung parenchyma by fibrosis with architectural distortion, honeycombing in a predominantly subpleural/paraseptal distribution, presence of fibroblastic foci, and absence of features suggesting an alternative diagnosis such as granulomas, organizing pneumonia, or hyaline membranes.
 HRCT has been shown in several clinical studies to be highly accurate for the presence of UIP pattern on SLB, with a positive predictive value as high as 90–100%.
HRCT has been shown in several clinical studies to be highly accurate for the presence of UIP pattern on SLB, with a positive predictive value as high as 90–100%.
 HRCT has become an essential tool in the diagnosis of IIP. Joint International Society guidelines for diagnosing IPF recommend against SLB if UIP pattern is present on HRCT, and alternative causes of ILDs have been excluded.
HRCT has become an essential tool in the diagnosis of IIP. Joint International Society guidelines for diagnosing IPF recommend against SLB if UIP pattern is present on HRCT, and alternative causes of ILDs have been excluded.
 In order for an HRCT to be consistent with the diagnosis of IPF, it has to demonstrate:
In order for an HRCT to be consistent with the diagnosis of IPF, it has to demonstrate:
1. reticular infiltrates in a predominantly basilar and subpleural distribution
2. honeycombing with/without traction bronchiectasis
3. an absence of features inconsistent with the diagnosis of UIP (e.g., ground glass, upper lung predominance, cysts, mosaic attenuation, and micronodules).
 For full details on the noninvasive diagnosis of IPF the reader is referred to the Official ATS/ERS/JRS/ALAT Statement on the diagnosis and treatment of IPF published in 2011.
For full details on the noninvasive diagnosis of IPF the reader is referred to the Official ATS/ERS/JRS/ALAT Statement on the diagnosis and treatment of IPF published in 2011.
• There is good circumstantial evidence that HRCT diagnosis is similarly accurate in other ILDs. In one study of patients with ILD in which the majority had a pre-existing histologic diagnosis, the correct first choice HRCT diagnosis was made in 87% of cases, with a remarkable level of agreement between the radiologic observers.
• HRCT appearance is highly suggestive of, or sometimes pathognomonic for:
 LAM
LAM
 LCH
LCH
 Pulmonary alveolar proteinosis (PAP)
Pulmonary alveolar proteinosis (PAP)
 HP
HP
• It is very important to note, however, that many studies of HRCT in ILD utilized experienced academic radiologists in making the diagnoses. The level of accuracy is often less in community settings. In these cases, or where the radiographic findings are equivocal, it may be necessary to move to a tissue diagnosis (see following sections).
• One of the great advantages of HRCT is the ability to detect coexisting pathology at the time of scanning.
• Once all coexisting conditions are diagnosed, management of patients with ILD can be optimized.
 One example is the coexistence of lung cancers and COPD in smokers with ILD.
One example is the coexistence of lung cancers and COPD in smokers with ILD.
 For example, PLCH, RB-ILD, and DIP occur almost exclusively in smokers. Therefore, all these patients are at higher risk of developing COPD and lung cancers, which can be detected at the time of scanning.
For example, PLCH, RB-ILD, and DIP occur almost exclusively in smokers. Therefore, all these patients are at higher risk of developing COPD and lung cancers, which can be detected at the time of scanning.
• Finally, although some clinicians believe serial CT evaluation is a valuable adjunct for monitoring disease progression in selected cases of ILD, the clinical utility of HRCT scanning in this capacity is overwhelmingly anecdotal and is in need of formal evaluation.
• As such, no firm recommendations regarding the monitoring of disease progression can be made at the present time.
Lung Sampling
Bronchoalveolar Lavage
• Bronchoalveolar lavage (BAL) is a method for sampling bronchial and alveolar epithelial secretions by instilling sterile saline into the distal lung units and retrieving the fluid for microscopic analysis.
• BAL is performed during fiberoptic bronchoscopy (FOB) and the target site is usually guided by HRCT.
• In general the procedure is well tolerated, but may carry an increased risk of complications in patients with severe hypoxemia or bleeding diathesis.
• The returned fluid carries a mixture of cellular and acellular components, pathogens, proteins, and insoluble particles that can be used for culture, cytology, and histologic analysis.
• The composition of BAL fluid may be diagnostic or suggestive of certain ILDs including:
 PAP (milky white fluid with positive periodic acid–Schiff staining)
PAP (milky white fluid with positive periodic acid–Schiff staining)
 acute or chronic eosinophilic pneumonia (>25% eosinophils in the cell differential)
acute or chronic eosinophilic pneumonia (>25% eosinophils in the cell differential)
 diffuse alveolar hemorrhage (increasingly bloody aliquots of aspirated fluid)
diffuse alveolar hemorrhage (increasingly bloody aliquots of aspirated fluid)
 malignancy (positive cytology)
malignancy (positive cytology)
 infection (positive bacterial, fungal, or viral cultures)
infection (positive bacterial, fungal, or viral cultures)
 pneumoconiosis (fluid contains asbestos bodies or silica)
pneumoconiosis (fluid contains asbestos bodies or silica)
 drug-induced pneumonitis or HP (>50% lymphocytes)
drug-induced pneumonitis or HP (>50% lymphocytes)
Transbronchial Lung Biopsy
• There is some controversy about the ideal lung biopsy technique for patients with ILD.
• Essentially, two forms of biopsy exist: transbronchial biopsy (TBBx) performed via FOB, and SLB.
• When deciding on a biopsy, a number of factors need to be taken into consideration.
• These factors include:
 the clinical condition of the patient
the clinical condition of the patient
 the skill of the surgeons or bronchoscopists
the skill of the surgeons or bronchoscopists
 the facilities available at the medical center concerned
the facilities available at the medical center concerned
 the disease process itself (this is very important)
the disease process itself (this is very important)
• The overall yield for TBBx in all forms of ILD is ∼50%.
• However, certain diseases are very amenable to diagnosis by TBBx.
• These diseases are predominantly bronchiolocentric (centered around the bronchioles) because the biopsy forceps must bite through the small peripheral bronchi to obtain lung tissue.
• Diseases with a high diagnostic yield on TBBx include:
 sarcoidosis (>95% in experienced hands)
sarcoidosis (>95% in experienced hands)
 berylliosis
berylliosis
 HP (subacute form)
HP (subacute form)
 PAP
PAP
 lymphangitic carcinomatosis
lymphangitic carcinomatosis
 bronchoalveolar carcinoma
bronchoalveolar carcinoma
• Conversely, TBBx cannot be used to diagnose some diseases owing to the small size of the tissue specimens.
• In these cases, TBBx may lead to an incorrect diagnosis being made.
• Examples include IPF and COP (also known as bronchiolitis obliterans organizing pneumonia).
 IPF has a diffuse and heterogeneous pathology with numerous pathologic features being required to confirm the diagnosis. Therefore, the findings of isolated parenchymal fibrosis on a TBBx are nonspecific and inadequate for the diagnosis of IPF.
IPF has a diffuse and heterogeneous pathology with numerous pathologic features being required to confirm the diagnosis. Therefore, the findings of isolated parenchymal fibrosis on a TBBx are nonspecific and inadequate for the diagnosis of IPF.
 COP is characterized by the plugging of bronchioles by growths of myxoid connective tissue, which occur diffusely throughout the lower zones of the lungs. However, localized injury (e.g., pneumonia) can result in the formation of identical lesions as part of a reparative response.
COP is characterized by the plugging of bronchioles by growths of myxoid connective tissue, which occur diffusely throughout the lower zones of the lungs. However, localized injury (e.g., pneumonia) can result in the formation of identical lesions as part of a reparative response.
• In addition to errors resulting from biopsy size, TBBx can yield poor results from sampling error (biopsying unaffected areas of lung), and crush artifact (crushing of the tissue by the biopsy forceps).
 To overcome these sampling errors and increase the TBBx yield, the lung is sampled multiple (10–20) times in different lobes.
To overcome these sampling errors and increase the TBBx yield, the lung is sampled multiple (10–20) times in different lobes.
 Yields are further increased by the use of HRCT to localize affected areas that are then targeted for biopsy.
Yields are further increased by the use of HRCT to localize affected areas that are then targeted for biopsy.
Surgical Lung Biopsy
• Indications for SLB include diseases such as vasculitis, NSIP, COP, DIP, and cases of suspected IPF in which HRCT findings are equivocal.
• SLB may be performed using two different techniques:
 video-assisted thoracoscopic surgery (VATS)
video-assisted thoracoscopic surgery (VATS)
 open thoracotomy
open thoracotomy
• VATS biopsy is less invasive than open lung biopsy obtained through an open thoracotomy.
• It results in similar diagnostic yields with less morbidity and shorter hospital stays.
• It is, however, unavailable in some centers and quite dependent on the skill of the surgeon.
• Similar to BAL and TBBx, SLB should be guided by HRCT to target areas of lung with abnormal findings.
• In general, obtaining biopsy samples from more than one lobe in a targeted fashion improves the diagnostic yield and accuracy.
• As with all biopsies, histopathologic data from SLBs should be correlated with a detailed clinical history, physical examination, and radiographic data to establish a diagnosis.
TREATMENT
General Treatment
• A comprehensive review of specific treatments for the various ILDs is well beyond the scope of this discussion.
• In general, if a causative agent is identified, withdrawal and avoidance of the offending agent (e.g., drugs, occupational exposures, and cigarette smoke) should be implemented immediately.
• Nonpharmacologic interventions such as pulmonary rehabilitation and supplemental oxygen should be initiated based on physiologic testing results to maintain functional status.
• Other comorbidities such as coronary artery disease, pulmonary hypertension, gastroesophageal reflux disease, and thromboembolic disease should be addressed as indicated.
• Treatment may involve both pharmacologic and nonpharmacologic therapies such as lung transplantation.
• For patients with progressive disease and severe physiologic impairment, who are ineligible for therapy or lung transplantation, end-of-life care issues should be addressed in the inpatient and outpatient setting.
Pharmacologic Treatment
• Pharmacologic treatment depends on the specific etiology of the ILD.
• In general collagen vascular, hypersensitivity, and autoimmune diseases are treated with glucocorticoids and/or immunosuppressive agents with varying degrees of success.
• Targeted therapies, such as granulocyte macrophage colony–stimulating factor (GM-CSF) for PAP may be considered.
• There have been promising advances in the field of LAM, which is now treated with sirolimus in selected cases. Other drugs for the treatment of LAM are currently in clinical trials.
• In 2014, two new drugs were approved by the FDA for treatment of IPF: pirfenidone and nintedanib. Both drugs were shown to slow the progression of disease, but did not reverse fibrosis. No significant mortality benefit has been demonstrated for either agent.
 Pirfenidone is an antifibrotic agent that reduces fibroblast proliferation, inhibits collagen production, and reduces production of fibrogenic mediators. Primary side effects include photosensitivity and GI upset.
Pirfenidone is an antifibrotic agent that reduces fibroblast proliferation, inhibits collagen production, and reduces production of fibrogenic mediators. Primary side effects include photosensitivity and GI upset.
 Nintedanib is a tyrosine–kinase inhibitor that targets vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR). Primary side effects include diarrhea, nausea, and other GI effects.
Nintedanib is a tyrosine–kinase inhibitor that targets vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR). Primary side effects include diarrhea, nausea, and other GI effects.
Lung Transplantation
• For selected patients without multiple comorbidities, lung transplantation may be an option.
• More detail on lung transplantation can be found in Chapter 29.
CONCLUSION
• ILD comprises a wide spectrum of diseases accounting for a considerable portion of everyday pulmonary practice.
• The pathogenesis of many of these diseases remains poorly understood and requires further investigation to facilitate development of novel therapies.
• Management of patients with ILD requires the clinician to integrate radiographic, physiologic, and histopathologic information with a detailed history and physical examination to make an accurate diagnosis and determine the optimal course of treatment.
• Many of these patients should be referred to clinical centers experienced in their treatment, or that offer clinical trials or lung transplant programs to appropriately facilitate their management.
REFERENCES
1. Deconinck B, Verschakelen J, Coolen J, et al. Diagnostic workup for diffuse parenchymal lung disease: schematic flowchart, literature review and pitfalls. Lung. 2012;191(1):19–25.
2. Meyer KC, Raghu G, Baughman RP, et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–14.
3. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
4. Wuyts WA, Agostini C, Antoniou K, et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2012;41(5):1207–18.
5. Ferguson EC, Berkowitz EA. Lung CT: Part 2, the interstitial pneumonias—clinical, histologic, and CT manifestations. AJR Am J Roentgenol. 2012;199(4):W464–76.
6. Swensen SJ, Aughenbaugh GL, Myers JL. Diffuse lung disease: diagnostic accuracy of CT in patients undergoing surgical biopsy of the lung. Radiology. 1997;205:229–34.
7. Garcia CK. Idiopathic pulmonary fibrosis. Update on genetic discoveries. Proc Am Thorac Soc. 2011; 8(2):158–62.
8. Kligerman SJ, Groshong S, Brown KK, et al. Nonspecific interstitial pneumonia: radiologic, clinical, and pathologic considerations. Radiographics. 2009;29(1):73–87.
9. Doyle TJ, Hunninghake GM, Rosas IO. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med. 2012;185(11):1147–53.
10. Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest. 2012;141(1):222–31.
11. Fischer A, du Bois R. Interstitial lung disease in connective tissue disorders. Lancet. 2012;380(9842):689–98.
12. Capobianco J, Gimberg A, Thompson BM, et al. Thoracic manifestations of collagen vascular diseases. Radiographics. 2012;32(1):33–50.
13. Selman M, Pardo A, King TE. Hypersensitivity Pneumonitis. Insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314–24.
14. Hirschmann JV, Pipavath SN, Godwin JD. Hypersensitivity pneumonitis: a historical, clinical and radiologic review. Radiographics. 2009;29(7):1921–38.
15. Chong S, Lee KS, Chung MJ, et al. Pneumoconiosis: comparison of imaging and pathologic findings. Radiographics. 2006;26(1):59–77.
16. Frankel SK, Cosgrove GP, Fischer A, et al. Update in the diagnosis and management of pulmonary vasculitis. Chest. 2006;129(2):452–65.
17. Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med. 1999;160(2): 736–55.
18. Criado E, Sanchez M, Ramirez J, et al. Pulmonary sarcoidosis: typical and atypical manifestations at high-resolution CT with pathologic correlation. Radiographics. 2010;30(6):1567–86.
19. Johnson SR, Cordier JF, Lazor R, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010;35(1):14–26.
20. Caminati A, Cavazza A, Sverzellati N, et al. An integrated approach in the diagnosis of smoking-related interstitial lung diseases. Eur Respir Rev. 2012;21(125):207–17.
21. Ghafoori P, Marks LB, Vujaskovic Z, et al. Radiation-induced lung injury. Assessment, management, and prevention. Oncology (Williston Park). 2008;22(1):37–47.
22. Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011; 20(120):98–107.
23. Raghu G, Brown KK. Interstitial lung disease: clinical evaluation and keys to an accurate diagnosis. Clin Chest Med. 2004;25(3):409–19.
24. Travis WD, Costabel U, Hansell DM, et al. An Official American Thoracic Society/European Respiratory Society Statement: update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


