Inflammatory Noninfectious Cardiovascular Diseases
Carolyn A. Altman
William B. Kyle
Kristen Sexson Tejtel
Marietta M. DeGuzman
Santiago Valdes
Andrea Ramirez
Introduction
The vasculitides are a heterogeneous group of systemic disorders characterized by inflammatory processes of the blood vessels leading to tissue injury. The site of vessel involvement, size of the affected vessels, extent of vascular injury, and underlying pathology determine the disease phenotype and disease severity. This chapter reviews those inflammatory, noninfectious diseases that most commonly affect the heart in the pediatric population (Kawasaki disease is discussed separately in Chapter 58).
Juvenile Idiopathic Arthritis
Juvenile idiopathic arthritis (JIA), formerly known as juvenile rheumatoid arthritis (JRA) and juvenile chronic arthritis (JCA), is the umbrella term used by the International League of Associations for Rheumatology (ILAR) to denote those childhood arthritides of unknown etiology which present before age 16 and endure at least 6 weeks (1,2,3,4,5). This is the classification system in most widespread use currently.
Six separate disease subtypes are described, with heterogeneous clinical, laboratory, genetic, and demographic features (3,5). These disease categories include:
Systemic arthritis
Polyarthritis
Oligoarthritis
Enthesitis-related arthritis
Psoriatic arthritis
A limitation of the ILAR classification for JIA is that the majority of studies and literature in juvenile arthritis predate ILAR criteria. The previous JRA nomenclature, used in many studies, defined three groups, including systemic JRA, oligoarticular JRA, and polyarticular JRA. Other studies may reference the JCA classification scheme developed by the European League Against Rheumatism (EULAR) (6,7)
Another limitation of the ILAR classification is that more than one disease is potentially lumped together within a single disease subtype (8). As well, although these diseases are theoretically not overlapping, there are children who meet inclusion or exclusion criteria for more than one category. Some children may fit better into a different category at an older age than the original category assigned at diagnosis (9). The classification of childhood arthritides is likely to continue to be refined and modified as genetic- and pathophysiology-based definitions for each disease evolve.
JIA Epidemiology
JIA Etiology/Pathophysiology/Diagnosis
The etiology of JIA is largely unknown but likely multifactorial given heterogeneity of the disease. The immune dysregulation in JIA is thought to be influenced by genetic and environmental triggers. Gender may also play a role since JIA tends to be more common in females.
The two subtypes associated with cardiac involvement are systemic and polyarthritis. They will be described in more depth here.
Systemic JIA
Systemic JIA is characterized by arthritis, daily intermittent fever of at least 2 weeks duration (quotidian for a minimum of 3 days), with at least one of the following features: evanescent, erythematous rash, generalized lymph node enlargement, hepatomegaly, splenomegaly, or serositis. Children with this subtype make up 5% to 15% of the cases of JIA (13). The sex ratio is about equal. A recent multicenter study revealed a peak between 1 and 5 years of age, however, other studies have shown that there is no definite peak incidence (14,15,16,17,18,19,20,21). Additional extra-articular findings may include cardiac involvement, pleural involvement, and nonspecific abdominal pain. Approximately 40% of these patients develop evidence of severe, progressive joint disease. The laboratory findings in the classic picture include anemia, leukocytosis, and an elevated sedimentation rate. Antinuclear antibodies (ANAs), rheumatoid factor (RF), and HLA-B27 are usually negative. Patients are at risk for developing macrophage activation syndrome (MAS) during disease flare which may cause a drop in two cell lines, hyperferritinemia, hypertriglyceridemia, hypofibrinogenemia, increased D-dimer, prolonged PT and PTT, elevated liver enzymes, elevated LDH, and a fall in ESR. Bone marrow aspiration reveals hemophagocytosis. MAS incurs increased risk of mortality, and may occur at presentation or during times of disease flare up (22,23).
Polyarthritis JIA
Polyarthritis JIA is characterized by the presence of arthritis in five or more joints, and occurs in approximately 15% to 25% of children with JIA (24,25). The age of disease onset varies based on the absence or presence of an RF. RF-negative JIA has a bimodal distribution occurring at ages 1 to 3 and 9 to 14 versus RF-positive JIA that mainly occurs between 10 and 13 years of age (1,26). Approximately 3% of children with JIA are RF-positive (13,27,28,29). Children who are RF-positive are more likely to develop severe chronic arthritis with erosive disease and worse prognosis than those who are RF-negative. The RF-positive subgroup closely resembles classic adult-onset rheumatoid arthritis. Other antibodies that may be present in both subgroups include anticyclic citrullinated peptide
(CCP) and ANAs. The presence of an ANA is as high as 57% in the RF-negative and 80% in RF-positive subgroups (30). Clinically, patients may experience other manifestations, including fatigue, growth issues, or uveitis. Rarely, patients may experience low-grade fever, subcutaneous nodules (typically noted over extensor surfaces of extremities), cardiovascular or pulmonary disease. Of note, cardiovascular and pulmonary involvement is less common in comparison to patients with systemic onset JIA.
(CCP) and ANAs. The presence of an ANA is as high as 57% in the RF-negative and 80% in RF-positive subgroups (30). Clinically, patients may experience other manifestations, including fatigue, growth issues, or uveitis. Rarely, patients may experience low-grade fever, subcutaneous nodules (typically noted over extensor surfaces of extremities), cardiovascular or pulmonary disease. Of note, cardiovascular and pulmonary involvement is less common in comparison to patients with systemic onset JIA.
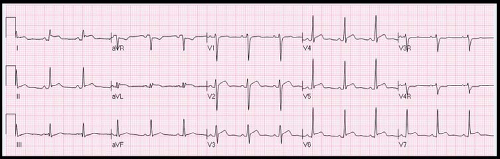 Figure 60.1 ECG in JIA patient with pericarditis demonstrating diffuse ST segment elevation. |
General Treatment Strategies for JIA
Pediatric rheumatology experts guide the general treatment for JIA, modulating therapy depending upon the subtype and clinical course. Generally, among the conventional disease-modifying antirheumatic drugs, methotrexate is the safe and effective first-line therapy in those with peripheral arthritis (3,27,31,32). Sulfasalazine is also used. Nonsteroidal anti-inflammatory drugs are not recommended as monotherapy for arthritis that persists for 2 months. Corticosteroids, both oral and intra-articular, continue to be used, but as adjunctive or bridge therapies (3,33,34,35). A variety of biologic agents have been reported as safe and effective in JIA. Routinely employed currently are tumor necrosis factor (TNF) inhibitors such as etanercept, monoclonal antibodies against TNF such as adalimumab and infliximab, and interleukin-1 or 6 inhibitors (3,5).
Cardiac Involvement in JIA
Cardiac involvement is the second major cause of mortality in this disease, thus it warrants careful evaluation and follow-up (6,7). JIA may affect the pericardium, myocardium, endocardium, and/or the autonomic regulation of heart rate and blood pressure. Obesity and abnormal lipid metabolism may also impact cardiovascular health.
Pericardium
Pericarditis is the most common cardiac finding in JIA (8,14). Clinical studies have reported that pericarditis occurs in 7% to 36% of patients, most commonly those with systemic JIA (9,16,36,37). However, most patients are only mildly symptomatic or have no symptoms at all. The lack of symptoms makes determining the overall incidence difficult to ascertain, but necropsy studies have found that 30% to 50% of JIA patients had some pericardial involvement (10,11). Symptoms of pericarditis are typical of pericarditis of any etiology, including acute substernal chest pain, which can be referred to the back, shoulder, or neck. Patients may be more comfortable sitting up or leaning forward, and experience worse pain and increased dyspnea while supine. Episodes of pericarditis simultaneously tend to occur with symptoms and signs of systemic JIA such as fevers, rash, and arthritis. Patients with systemic disease are also more likely to have larger effusions compared to those with nonsystemic disease (12,38). Patients with moderate-to-large effusions are more likely to be symptomatic compared to those with small effusions. Tachycardia and friction rubs are the most common physical examination findings of pericarditis.
Diagnostic evaluation may include a chest radiograph to assess for cardiomegaly, although this requires a large pericardial effusion to be present. Electrocardiography (ECG) evidence of pericarditis includes PR depression, diffuse ST segment elevation and T wave inversion (Fig. 60.1). Decreased voltages and electrical alternans can be seen in the setting of large effusions. Echocardiography is instrumental in demonstrating the size and assessing the hemodynamic significance of an effusion (Fig. 60.2). However, patients
may have chest pain and ECG findings diagnostic of pericarditis with trivial or even no effusion. Fortunately, most JIA patients with pericarditis tend to have an insidious and slowly progressive course. Therefore, cardiac tamponade is a rare occurrence in children with JIA (10,13,39). The diagnosis of tamponade is made clinically in the patient with sustained sinus tachycardia, elevated jugular venous distention, pulsus paradoxus, and eventually the onset of hypotension secondary to poor cardiac output. (see Chapter 61 on Pericardial Disease).
may have chest pain and ECG findings diagnostic of pericarditis with trivial or even no effusion. Fortunately, most JIA patients with pericarditis tend to have an insidious and slowly progressive course. Therefore, cardiac tamponade is a rare occurrence in children with JIA (10,13,39). The diagnosis of tamponade is made clinically in the patient with sustained sinus tachycardia, elevated jugular venous distention, pulsus paradoxus, and eventually the onset of hypotension secondary to poor cardiac output. (see Chapter 61 on Pericardial Disease).
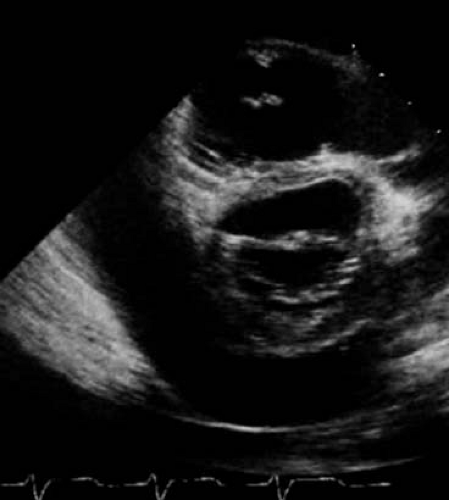 Figure 60.2 Pericardial effusion in JIA patient: echo parasternal short-axis view showing moderate posterior effusion. |
Myocardium
Myocarditis is reported to occur in 1.2% to 10% inpatients with JIA. Patients may present with isolated myocarditis or with myocarditis in association with pericarditis (14,15,16,17,18,19,20,21). Clinical presentation will depend on the severity of myocarditis, but symptoms may include tachycardia and dyspnea. If cardiac output is significantly compromised, hypotension, pulmonary edema, ascites, and lower extremity edema may develop. Chest radiograph may demonstrate cardiomegaly. ECG may show nonspecific ST and T wave changes, conduction abnormalities including PR prolongation and new bundle branch block. Cardiac arrhythmias (atrial or ventricular) can develop. Mildly elevated troponin levels may occur with myocarditis. Echocardiography is key in demonstrating ventricular dysfunction. Cardiac MRI can demonstrate evidence of ventricular dysfunction as well as areas of patchy myocardial fibrosis on delayed enhancement. Cardiac catheterizations are only very rarely performed now to obtain biopsies for histopathologic evaluation or viral PCR studies. Outside of the setting of acute myopericarditis, echocardiographic studies have shown mixed results in the evaluation of the myocardium in JIA patients. Some studies have shown lower ejection fractions (EFs) and shortening fractions (SF) compared to controls (although still within normal range), while others have not described any differences (14,22,40,41). LV end-diastolic and systolic dilation has been reported as well. In examining diastolic parameters, JIA patients have lower E waves and higher A waves, and, thus, lower E/A ratios and longer isovolumic relaxation times (IVRT) (14,24,36,41,42). These diastolic changes are similar to those found in patients with cardiomyopathies, hypertension, and ischemic heart disease. Diastolic dysfunction has been reported to increase in JIA patients with increased duration of disease, but the etiology of the dysfunction is not clear (25,41,43).
Endothelium
Endocardial involvement is relatively rare in JIA patients. The aortic and mitral valves are most commonly affected (18,36,37,44,45). Up to 25% of patients may demonstrate evidence of mitral thickening and insufficiency. Aortic involvement is less common, occurring in 5% to 10% of patients and limited typically to cusp thickening without obstruction or significant regurgitation (18,36,37,44,45).
Treatment of Myopericardial or Endocardial Involvement
Therapy for JIA patients with cardiac involvement is usually geared toward the underlying systemic inflammation and is guided by an expert in pediatric rheumatology. Management for pericarditis with tamponade includes the infusion of intravenous fluids until urgent pericardiocentesis can be accomplished. Management of heart failure is typical and includes angiotensin-converting enzyme inhibitors, beta-blockers, fluid restriction, diuretics, and possibly inotropic agents.
Heart rate, Blood Pressure, and Autonomic Function
Pediatric patients with JIA have higher heart rate and resting systolic and diastolic blood pressures compared to controls, although often still within the normal range (14,36). As hypertension constitutes a common cause of morbidity in adult JIA patients, close monitoring of blood pressure in affected children over time is important. Cardiac autonomic dysfunction may occur in patients with inflammatory diseases such as JIA. Symptoms can include palpitations, orthostatic intolerance (postural weakness, dizziness, lightheadedness, and syncope), and exercise intolerance. In a study of JIA patients, symptoms of cardiac autonomic dysfunction were found in 30% of patients (46).
Evaluation for autonomic dysfunction by noninvasive cardiovascular reflex autonomic function tests (AFTs) includes assessing resting heart rate, heart rate variability, heart rate response to Valsalva maneuver, and heart rate and blood pressure response to standing. In one study, as assessed by AFTs, 40% of JIA patients had evidence of cardiac autonomic dysfunction (46). Autonomic dysfunction was categorized as mild in 10%, and severe in 15%. Interestingly, 14.3% of JIA patients with cardiac autonomic dysfunction by AFTs had no symptoms.
Measurement of levels of neuropeptides involved in autonomic neural control of cardiovascular function may provide further useful information. Neuropeptide Y (NPY) and vasoactive intestinal peptide (VIP) levels have been reported as significantly lower compared to controls in JIA patients (46). As well, JIA patients with clinical evidence of cardiac autonomic dysfunction have lower levels of NPY and VIP compared to JIA patients with no clinical evidence of dysfunction. Serum VIP had a higher sensitivity and specificity for cardiac autonomic dysfunction than serum NPY (46).
JIA patients should be routinely evaluated for symptoms of autonomic dysfunction. Consideration should be given to performing AFTs and autonomic neuropeptide monitoring, as some patients will have subclinical disease. For those with evidence of disease, treatment can include increase in water and salt intake, wearing of lower extremity compression stockings, and exercise training. Pharmacologic therapy with beta-blockers can be added for those with symptomatic tachycardia. Studies are needed to evaluate long-term results of the above therapies in JIA patients (34,35,46).
Atherosclerosis
Patients with JIA typically have higher body mass index, fat percentages, and truncal fat than age-matched controls (47). Autopsy studies have shown that 30% of children with JIA have evidence of atherosclerosis (48). A typical lipid profile has not been elucidated (49). There are three major vascular markers that have been validated as measures of early atherosclerosis; flow-mediated dilation (FMD), carotid intima-media thickness (CIMT), and pulse wave velocity (PWV) (49). When evaluating these in JIA patients, however, the results have been mixed and contradictory secondary to the small and heterogeneous studies that are available. Further larger-scale studies are needed, particularly in those with systemic JIA who are likely to be at higher risk with higher levels of inflammation and long-term corticosteroid use (49).
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a chronic episodic multisystem disease with a wide range of manifestations. The Latin name “lupus,” which translates to “wolf” in English, was given because the skin manifestations resembled the bite of an animal (50). It was Sir William Osler who recognized the involvement of other organ systems, including the heart, and changed the name to its modern form. Libman and Sacks first described the eponymous “verrucous” endocarditis lesions in the early 20th century (51). Cardiac disease is the third leading cause of death in SLE behind renal disease and infection, with the incidence of cardiac disease increasing in patients with a longer course of disease (52).
SLE Epidemiology
Presentation before 18 years old occurs in about 15% of SLE cases, with an incidence between 0.36 and 0.9 per 100,000 children (53,54). Most affected children are between 12 and 16 years of age, with disease rarely seen before age 5 (53). Earlier age at onset (particularly prepubertal) is associated with more severe symptoms at onset, increased lifetime disease burden, and worse outcome (55). Overall, affected females outnumber affected males 8:1, but there is less gender disparity at younger ages.
SLE Etiology
SLE is often considered the prototype systemic autoimmune disease. It is likely that genetic susceptibility and environmental influences combine to produce the phenotype. Dysfunction within the innate and adaptive immune systems (including B- and T-cell dysregulation, immune complex deposition, complement activation and other factors) results in the loss of tolerance to self-antigens (56,57). ANAs (e.g., anti–double-stranded DNA) are commonly found in SLE patients. The presence of antiphospholipid antibody is clinically important as this increases potential for thrombosis. Exposure to sunlight, infections, drugs, and chemicals have been shown to play an important role in disease manifestation and course.
SLE Clinical Features and Investigation
The diagnosis of juvenile SLE is based on clinical, laboratory, and immunoserologic features. The most widely used diagnostic criteria for all ages have been those defined by the American College of Rheumatology (ACR) in 1982 with modification in 1997 (Table 60.1) (58,59,60). The clinical criteria highlight the multiorgan involvement of the disease. A person is defined as having SLE if any 4 or more of the 11 criteria are present, serially or simultaneously, during any interval of observation. However, these criteria were primarily developed in adult populations with little validation in pediatrics (61,62). Alternative criteria systems for the diagnosis of SLE include the Boston weighted (BW) criteria and the Systemic Lupus International Collaborating Clinics criteria (SLICC) (60,63,64,65,66). The SLICC criteria modified and expanded various elements of the ACR, and required 4 of 17 criteria to be met for the diagnosis, including 1 clinical and 1 immunologic. Fonseca et al. recently compared these 3 diagnostic criteria systems in a cohort of 81 juvenile SLE patients (62,67). They reported that the SLICC criteria had the best sensitivity and accuracy when looking at the first visit and first-year follow-up (62,68).
In juvenile SLE, more common features are nonspecific symptoms such as fatigue, fever, and weight loss. Other findings include gastrointestinal disease (hepatosplenomegaly, pancreatitis, and abdominal pain), neuropathies, pleural disease, conjunctivitis, and lymphadenopathy in addition to the cardiac manifestations detailed below. Symptoms most frequently encountered in the child and adolescent age range are those involving the pleura and pericardium, joints, kidneys, and skin.
SLE Cardiac Involvement
Just as SLE is expressed broadly throughout the body, multiple components within the cardiovascular system may be compromised, including pericardium, myocardium, endocardium, conduction tissue, and coronary arteries. Systemic and pulmonary hypertension contributes their own detrimental effects as do side effects of therapies necessary to ameliorate inflammation.
Cardiac involvement has been reported in 23% to over 50% of SLE patients. The wide range is due in part to which findings are classified as cardiac (e.g., systemic hypertension) and whether subclinical manifestations are included (versus only those that caused symptoms), as well as the sensitivity of the diagnostic test (imaging studies vs. autopsy). In a study reviewing 50 children with SLE, 68% had an abnormality on echocardiograms, most of which had been obtained routinely. All 10 patients whose echocardiograms were prompted by concerning symptoms had abnormal studies. While only 6% of the abnormalities were considered severe, the severity of echocardiographic findings did not correlate with other markers of disease severity (69,70,71,72).
TABLE 60.1 Abbreviated Summary of Clinical Criteria of Systemic Lupus Erythematosus, with 4 of 11 Criteria Required to Diagnose SLE per the 1997 Revision of the Original 1982 Criteria | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Pericardium
Pericardial disease is the most common cardiac manifestation of SLE. Pericarditis occurs in approximately 16% to 38% of all children with SLE (53,73,74,75,76). On autopsy, the incidence of pericardial disease is substantially higher than that which is detected clinically (64% vs. approximately 25%, respectively) (69,77). Histopathologically, the pericardium exhibits evidence of chronic and fibrinous pericarditis. Immune complex aggregate deposition in the pericardium is thought to mediate pericardial disease, correlating with clinical or histopathologic disease (78,79). Immune complexes are predominately identified in the perivascular portions of the pericardium and other heart tissues, deposited in a fine granular pattern. Not surprisingly, this mechanism of disease is similar to that found in other affected organs as well (79,80). Pericardial fluid is not routinely collected, but analysis demonstrates an exudate with a high protein concentration and normal to low glucose levels (81,82). The leukocyte count is elevated with a neutrophil predominance, and autoantibodies such as anti-DNA and ANA can be found (68,83).
Clinically, pericardial disease presents in the typical manner. Patients report sharp precordial chest pain and dyspnea. The pain may be pleuritic in nature or the pleura are commonly affected as part of a general serositis. Discomfort is often relieved by leaning forward. Signs include tachycardia, fever, and a pericardial rub. Large pericardial effusions may manifest with distant heart sounds and orthopnea. Jugular venous distention and pulsus paradoxus can occur in cardiac tamponade. However, as evidenced by the increased diagnosis on autopsy, pericardial involvement in this disease is often clinically silent.
ECG findings include sinus tachycardia, diffuse ST segment elevation not following a typical coronary artery distribution and PR segment depression (see Fig. 60.1 for ECG findings pericarditis). Large pericardial effusions may result in diminished QRS voltages and electrical alternans. An enlarged cardiac silhouette on chest x-ray supports the presence of a large pericardial effusion, but a normal heart size does not exclude pericarditis. Common echocardiographic findings include pericardial thickening and effusions. Cardiac tamponade is a rare phenomenon, with less than 1% of patients found to have tamponade in a review of >1,300 patients with SLE (84,85). Despite the frequency of histopathologic changes on autopsy, constrictive pericardial disease is also felt to be a rare complication in SLE, and data are limited to case reports.
Management for pericardial disease is dictated by the severity at presentation. Incidental, small pericardial effusions can be monitored expectantly while nonsteroidal anti-inflammatory medications are often the first-line treatment for pericarditis or more significant effusions. Steroid pulses are employed, often intravenously, for severe forms of the disease or when effusions fail to respond to initial therapy. Finally, when pericardial involvement is severe or recurrent, it is important to optimize systemic therapy for SLE. Pericardiocentesis is rarely necessary but should be used in cases of cardiac tamponade. It is not routinely indicated for diagnostic purposes.
Endocardium
Valvular heart disease is an important cardiac manifestation of SLE. Reported prevalence of valvular disease in SLE ranges from 18% to 60% on echocardiographic studies, and higher in autopsy specimens (81,84,86,87,88,89,90). Left-sided valves are most often affected, with the mitral valve affected more than the aortic. Right-sided involvement in multi-valve disease can occur. Common manifestations, in order of frequency, include valve thickening, verrucous endocarditis (classic Libman–Sacks lesions), regurgitation, and stenosis (72,91). Thickening of the valves is typically diffuse and involvement of the annulus and subvalvar apparatus is not uncommon (Fig. 60.3). Chordae can become fibrotic and scarred, but chordal rupture is rare. Studies assessing valve function over time in pediatrics are lacking. However, in a study of 69 adults with SLE, of the 25% with valve regurgitation on echocardiogram, 65% had at least moderate regurgitation. When reimaged at a mean interval of about 2 years, most of the valve lesions remained stable. However, there was a significant minority of lesions that either resolved or evolved in that time frame (17% and 12%, respectively), likely owing to the dynamic nature of the inflammatory process (72,92).
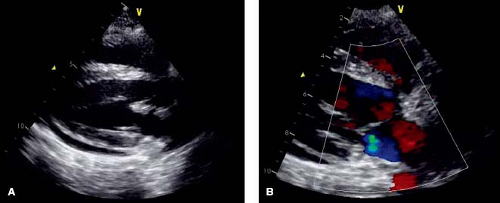 Figure 60.3 Parasternal long-axis echocardiogram demonstrating thickening of the anterior mitral valve leaflet with upper mild regurgitation and a small pericardial effusion in a 15-year-old female with SLE. |
Microscopically, SLE-related valve lesions contain deposits of anticardiolipin antibodies and complement components. The valves are thickened, calcific, and edematous with focal surface irregularities and cellular infiltration (93,94). The relationship between antiphospholipid antibodies and valvular heart disease remains an area of active investigation with some studies demonstrating a higher incidence of lesions in patients with antiphospholipid antibodies (95,96). Deposition of thrombotic material leading to commissural fusion is another proposed mechanism for valve dysfunction (79,87).
The verrucous, nonbacterial endocarditis described by Libman and Sacks is clearly the most well-recognized and well-described valve abnormality even though it may be clinically less important than the valve lesions described above. The lesions are usually small (1 to 4 mm) nodules composed of fibrin with proliferating and degenerating cells along with variable amounts of inflammatory cells (69,97). Libman–Sacks lesions are found in about 50% of autopsy specimens, but are usually too small to be visualized reliably on echo. They are usually located either along the annular surface of the valve in the pocket created where the leaflet meets the surrounding structures, or on the edge of the leaflet itself. Verrucae can also be found on either surface of the valve (or valves), at the commissures, on the chordae tendineae, on the papillary muscles, or even on the atrial or ventricular endocardium. When they can be visualized by echo, they are difficult to distinguish from bacterial vegetations. Clinical correlation with blood cultures and other markers of infection are necessary as infective endocarditis is a known source of morbidity in SLE patients who require chronic immunosuppression for disease control (69,72,98). Libman–Sacks nodules are usually benign, but may result in valve insufficiency or stenosis. An uncommon but concerning complication is an embolic event, with ischemic stroke or peripheral embolization of the nodule itself or an associated thrombus, in this population predisposed to hypercoagulation (61,99,100).
Management for Valvular Disease
Routine echocardiographic surveillance in patients with SLE is warranted, particularly in the presence of antiphospholipid antibody syndrome, interstitial lung disease, or pulmonary hypertension.
However, needed imaging frequency has not been clearly defined and depends on the clinical features and course of the disease. The majority of valve manifestations of SLE can be managed expectantly. A new echocardiographic finding does not necessarily mandate a change in therapy, as valve disease is not always directly associated with a change in disease activity. However, optimization of anti-inflammatory, immunolytic, and anticoagulation therapy is prudent (72,101). Surgical intervention may be necessary for hemodynamically significant lesions that are unresponsive to medical therapy. Operative experience suggests that valve repair may be less durable in SLE patients (63,65,102). When bioprosthetic valves are used in replacing a diseased valve, there is concern for valvulitis and accelerated degeneration of the replacement (66,103). The use of a mechanical valve mitigates this possibility but brings with it the difficult issue of anticoagulation management. After a recent successful case report, the role of transcatheter aortic valve replacement in adult SLE patients with valve dysfunction remains to be defined (67,99). With improvements in therapy and longevity in patients with this disease, valvular disease will remain an important consideration.
However, needed imaging frequency has not been clearly defined and depends on the clinical features and course of the disease. The majority of valve manifestations of SLE can be managed expectantly. A new echocardiographic finding does not necessarily mandate a change in therapy, as valve disease is not always directly associated with a change in disease activity. However, optimization of anti-inflammatory, immunolytic, and anticoagulation therapy is prudent (72,101). Surgical intervention may be necessary for hemodynamically significant lesions that are unresponsive to medical therapy. Operative experience suggests that valve repair may be less durable in SLE patients (63,65,102). When bioprosthetic valves are used in replacing a diseased valve, there is concern for valvulitis and accelerated degeneration of the replacement (66,103). The use of a mechanical valve mitigates this possibility but brings with it the difficult issue of anticoagulation management. After a recent successful case report, the role of transcatheter aortic valve replacement in adult SLE patients with valve dysfunction remains to be defined (67,99). With improvements in therapy and longevity in patients with this disease, valvular disease will remain an important consideration.
Coronary Artery Disease
Coronary artery disease in SLE impacts morbidity and mortality in the pediatric and young adult population, and especially in young women (68,104). Women 35 to 44 years of age have been found to be at over 50 times greater risk of having a myocardial infarction (MI) than their non-SLE counterparts (105,106). Pediatric-onset SLE has been found to be an independent risk factor for future MI. In one study, the average age of first MI was 32 years (107,108). MI has been reported in SLE patients as young as 5 years (106,109).
In a review of published literature in patients 35 years of age or younger with coronary artery disease, MI was the most common initial cardiac manifestation (91%) followed by congestive heart failure (27%), and then sudden death (12%) (69,110). Based on angiography or autopsy, atherosclerosis was the most common coronary artery lesion in SLE followed by arteritis (69,111). The left anterior descending artery was the most commonly affected vessel, and about half of the patients had thrombosis in the coronary artery at the time of evaluation. Lupus arteritis can lead to the development of aneurysms or vasospasm of the coronary arteries.
Antiphospholipid syndrome (APS) is an important condition that may occur in rheumatic diseases like SLE. Its extreme manifestation, the so-called “catastrophic APS,” results in ischemia in multiple organs and is often fatal. Miller et al. reported on a fatal MI in an 8-year-old African-American girl with a 5-month history of SLE and CAPS (74,112). At autopsy, transmural ischemia associated with coronary arteriopathy and an acute thrombus was found. The authors noted the absence of inflammatory or atherosclerotic changes in the coronary arteries, speculating that the thrombogenic potential of APS led to the thrombosis.
Myocardial perfusion can be abnormal in SLE patients even without cardiac symptoms. In a study of 40 such children (10 to 20 years old), defects on thallium myocardial perfusion scans were demonstrated in 16% (75,113).
Similar to other phenotypic expressions of SLE, immune complex deposition is implicated in the development of coronary artery disease. Coronary artery intimal damage hastens the development of atherosclerosis. The contribution of antiphospholipid antibodies, including anticardiolipin antibody, to coronary artery disease is a topic of debate. There are studies that report an association between the level of these antibodies and coronary artery disease and others reporting no association. One study reported that CIMT was greater in adolescents with SLE compared to healthy controls, and, within the SLE cohort, findings were related to the presence of nephrotic-range proteinuria (40,77). This supports the idea that disease severity perhaps plays more of a role than duration. Independent of therapy, SLE patients have lower HDL levels at baseline (42,78). Hypertriglyceridemia and hypercoagulability in SLE are other risk factors contributing to coronary artery disease. Risk factors for increased carotid intimal thickness in children with SLE includes the traditional risk factors such as increased BMI, male gender, and increasing age, as well as the use of azathioprine and prednisone (43,80). The role of steroids in this process is interesting. On one hand, steroids, as a mainstay of therapy, improve longevity; however, the side-effect profile of this medication is quite deleterious with respect to the coronary arteries. Well-known side effects include hypertension, hyperlipidemia, weight gain, and steroid-induced diabetes mellitus (82,114).
Management for Coronary Artery Disease
Screening for hyperlipidemias should be part of the routine management of pediatric SLE patients per the AHA guidelines on Cardiovascular Risk Reduction in High-Risk Pediatric Patients, where SLE is considered a Tier II, or moderate risk, disease (68,115). Exercise testing, nuclear perfusion scans, and carotid ultrasound may also be employed in the assessment of cardiac and vascular function.
Presentation of MI should be suspected in SLE patients with the familiar symptoms of chest pain or pressure, dyspnea, weakness, sweating, nausea, and vomiting. However, absent the classic presentation, chest pain in children with SLE can present a diagnostic dilemma. While pericarditis is clearly the more common cause of chest pain in the pediatric age group and the SLE population in general, one should avoid ascribing the pain to pericarditis without considering infarction in the differential. Providers should have a high index of suspicion, given the increased rate of cardiovascular events even in the absence of traditional risk factors (28,84,109,116,117,118,119). An ECG and quantification of serum cardiac enzymes should be readily employed for chest pain or any other concerns for ischemia. Echocardiography can help in the evaluation by assessment of regional wall motion abnormalities, global ventricular dysfunction, or pericardial effusion (Video 60.1). More involved and invasive tests such as computed tomography and cardiac catheterization with angiography may be considered necessary depending upon the clinical scenario and preceding workup. Cardiac MR may demonstrate scarring related to MI (Fig. 60.4). Medical management of acute MI involves increasing myocardial oxygen
delivery while decreasing demand. This can be accomplished using a combination of supplemental oxygen, antiplatelet drugs, anticoagulants, antianginal drugs (including beta-blockers and nitrates), analgesics, and anti-inflammatory agents such as HMG-CoA inhibitors (statins). Catheterization with stent placement may be necessary in certain situations including the presence of an ST elevation MI, and consultation with experts in ischemic heart disease is advised (see Chapter 63).
delivery while decreasing demand. This can be accomplished using a combination of supplemental oxygen, antiplatelet drugs, anticoagulants, antianginal drugs (including beta-blockers and nitrates), analgesics, and anti-inflammatory agents such as HMG-CoA inhibitors (statins). Catheterization with stent placement may be necessary in certain situations including the presence of an ST elevation MI, and consultation with experts in ischemic heart disease is advised (see Chapter 63).
 Figure 60.4 MR showing scarring of the LV apex via gadolinium-delayed enhancement in a childhood lupus patient. |
Treatment of coronary arteritis as a manifestation of an SLE flare requires consultation with pediatric rheumatology experts, and may include escalation or addition of steroid therapy and higher level of immunomodulation (e.g., rituximab, cyclophosphamide, mycophenolate mofetil); IVIG and biologic agents may be utilized as well. Prevention or minimizing the impact of atherosclerotic disease requires taking appropriate preventive measures early in childhood. Paramount to this goal is the judicious use of steroids to achieve therapeutic objectives while minimizing side effects. Hydroxychloroquine, another backbone of clinical therapy in SLE, has actually been shown to improve the lipid profile in SLE patients (27,28,29,86). Aggressive management of modifiable risk factors includes the familiar advisement of proper diet, exercise (60 minutes of aerobic activity per day in children and adolescents), and avoidance of smoking (84,120). A 6- to 12-month trial of diet and exercise therapy is warranted in most cases of dyslipidemia. Aggressive medical treatment of hyperlipidemia with statins is an intuitively attractive strategy, however, in a randomized placebo-controlled trial in adults (mean age 45 years) without clinical cardiovascular disease, 40 mg of atorvastatin daily did not have a significant impact on coronary artery calcium (detected by computed tomography), CIMT, or carotid plaque (3,27,31,32,88). Markers of inflammation and endothelial cell activity and measures of disease activity were similarly unaffected. The lack of efficacy data has led some to promote use of statins when patients meet routine indications and not empirically in the asymptomatic individual. There are other less well-tolerated (niacin, fibrates, bile acid sequestrants) and complementary (fish oil, garlic, antioxidant vitamins) therapies that play a smaller role in disease management (3,33,86).
Myocardium
Myocardial involvement in SLE can include myocarditis or the development of a cardiomyopathy with contributions from coronary pathology, myocardial inflammation, and/or as a side effect of hydroxychloroquine therapy.
Myocarditis is often clinically silent, with about 5% to 10% of SLE patients exhibiting symptoms (28,34,35,91,92,121,122,123). The diagnosis is made more frequently on autopsy with an incidence of 40% to 70% (93,95,118,124,125). Autopsy specimens demonstrate a diffuse granular deposition pattern of immune complex aggregates, especially in the vascular walls supplying the myocardium (27,28,31,79,118). The abundance of immune complex deposition has been positively associated with clinical and serologic evidence of disease severity.
When symptomatic, myocarditis presents in the typical manner with fever, tachycardia, and shortness of breath. The patient may experience chest pain, palpitations, or exercise intolerance. On examination, one may note a gallop rhythm or a new murmur, jugular venous distention or peripheral edema. While the incidence is low, myocarditis can be life threatening. In addition to ventricular dysfunction (which can occur acutely or chronically), heart block and lethal arrhythmias such as ventricular tachycardia and ventricular fibrillation do occur. Given the broad spectrum of potential cardiac involvement in SLE, heart failure from myocarditis must be distinguished from other manifestations of SLE, including coronary artery disease, hypertensive heart disease, and primary arrhythmias (27,28,32,97).
Diagnostic evaluation for myocarditis would include a chest x-ray, which may be normal or could demonstrate cardiomegaly and pulmonary venous congestion. Electrocardiographic findings are often nonspecific in myocarditis and include sinus tachycardia and diminished QRS voltages. T waves can be flat or inverted with or without ST segment changes. ECG should be carefully examined for signs of ischemia which may point to coronary involvement or, if diffuse, more severe global myocarditis. Suspicion of myocarditis should prompt an echocardiogram to evaluate ventricular function, valve regurgitation, and pericardial effusion. Like MI, regional wall motion abnormalities and an elevation of serum cardiac enzymes can be seen in myocarditis although they are typically less marked. Concentrations of inflammatory markers (white blood cell count, erythrocyte sedimentation rate, C-reactive protein) and B-type natriuretic peptide can be helpful, especially in trending response to therapy. Magnetic resonance imaging may be helpful in assessing for contrast enhancement. Myocardial biopsy is not sensitive and is infrequently performed.
Management of Myocarditis in SLE
High-dose pulse corticosteroids guided by pediatric rheumatology specialist are the mainstay of therapy. Optimization of the chronic treatment regimen then follows. Management of heart failure symptoms includes angiotensin-converting enzyme inhibitors, beta-blockers, fluid restriction, and diuretics. Inotropic or antiarrhythmic agents may be necessary. Prompt diagnosis and treatment maximize the chances of a favorable outcome.
Neonatal Lupus Erythematosus, Arrhythmias, Conduction System Disease
Congenital complete atrioventricular block (CCAVB) is the manifestation of neonatal lupus erythematosus (NLE) most familiar to pediatric cardiologists. CCAVB typically develops in the fetus between 16 and 28 weeks of gestation (31,98) (see Chapter 5 for discussion of Fetal Diagnosis and Management). In pregnancies of mothers with known positive SSA/SSB antibodies, with no previously affected children, between 1% and 5% of fetuses develop AVB (99,100,101,102,122,123,126,127). With a previous child with AVB or neonatal lupus, the risk increases strikingly to 11% to 19%. Other electrocardiographic findings of neonatal lupus include sinus bradycardia and QT prolongation.
Ventricular dysfunction and dilated cardiomyopathy may also develop in utero, within the first few weeks of life, or years later (33,103). The estimated incidence of dysfunction is 5% to 11% in patients with CCAVB. In fetuses and neonates with or without conduction system disease, endomyocardial involvement may include mitral or tricuspid valvulitis, pericarditis, myocardial dysfunction, or endocardial fibroelastosis (EFE) (28,29,53,54,99,104,117,118,119,128).
The etiology of the development of CCAVB is not entirely known. Maternal IgG SS-A/Ro and SS-B/La antibodies are transplacentally delivered to the fetus (106,108,127,129,130). One proposed mechanism is that the antibodies bind to myocytes causing apoptosis, triggering molecular mechanisms which inhibit removal of apoptotic cells, suppress protective cellular responses, and promote scarring (29,106). This would then result in the inflammation, calcification, fibrosis, and pathway interruption that is observed in the cardiac tissue, especially the AV node and the distal conduction system. Another hypothesis is that the antibodies from the mother act on L-type calcium channels in such a way as to dysregulate calcium homeostasis leading to downstream conduction deficits (56,57,110,130). Because not every fetus of a mother with the offending antibodies actually expresses the disease, genetic factors may be at play, including variability within the fetal major histocompatibility complex profile. Differences in protein expression throughout gestation are postulated to explain why a neonate may exhibit complete heart block when the mother is completely asymptomatic.
Fetal and neonatal cardiac manifestations of NLE incur significant morbidity and mortality. A European multinational retrospective study of fetuses and children with second- and third-degree
AVB found that intrauterine mortality was higher in fetuses with earlier gestational age at diagnosis, hydrops, depressed left ventricular function, and ventricular rate ≤50 beats per minute (111,127). In a study of 325 fetuses and newborns with cardiac NLE, there was a 17.5% overall mortality, with 6% of deaths in utero (28,131) Of live-born infants, the large majority of attrition occurred within the first year of life with a cumulative survival probability of 86% at 10 years of age. Half the children required a pacemaker in their first year of life, and about 70% were paced by 10 years of age.
AVB found that intrauterine mortality was higher in fetuses with earlier gestational age at diagnosis, hydrops, depressed left ventricular function, and ventricular rate ≤50 beats per minute (111,127). In a study of 325 fetuses and newborns with cardiac NLE, there was a 17.5% overall mortality, with 6% of deaths in utero (28,131) Of live-born infants, the large majority of attrition occurred within the first year of life with a cumulative survival probability of 86% at 10 years of age. Half the children required a pacemaker in their first year of life, and about 70% were paced by 10 years of age.
In individuals spared from the fetal, newborn, and early childhood complications described above, arrhythmias and conduction disturbances in SLE are a much more minor source of morbidity. Sinus tachycardia is common in adults, but the incidence of other arrhythmias is probably less than 10% (112,116). Atrial arrhythmias (premature atrial contractions, atrial flutter) are more common than ventricular arrhythmias and conduction defects. AV block (first, second, and rarely third degree), intraventricular conduction delay, and bundle branch block are encountered with an incidence of only 5% to 18% in adults (28,34,40,58,113,122,132,133). Corrected QT prolongation can be seen in patients with circulating autoantibodies. Arrhythmias, often transient, can be caused by an active inflammatory process (pericarditis, myocarditis) as well as acute or ongoing ischemia, and one should have a higher level of suspicion for arrhythmia when there is an acute disease flare. The mechanism of conduction deficits is likely related to inflammation, degeneration, and fibrosis found within the conduction tissue (42,127). Long-term use of hydroxychloroquine, common in SLE patients, is associated with cardiomyopathy and has been implicated in causing AV block, but this side effect is debated (28,43,114,115,134). When the presence of tachycardia, palpitations, or syncope is elicited on history, appropriate follow-up diagnostic testing such as an ECG and assessment of hematocrit, electrolyte, and thyroid hormone levels may be appropriate. Portable rhythm monitoring or exercise testing can be pursued depending on the index of suspicion.
Takayasu Arteritis
Introduction
Takayasu arteritis (TA) is the most common chronic, granulomatous inflammation of large vessels, affecting the aorta, its major branches, and the pulmonary arteries (28,109,116,117,118,119,122). Historically, TA has been called “pulseless disease,” the result of the progressive segmental vessel narrowing and occlusion which develop over time (27,28,29,120,135).
TA Diagnosis
EULAR/PRINTO/PRES provides criteria for the diagnosis of TA (3,27,29,31,32). A required feature is disease of the aorta or its major branches or the pulmonary artery (with no other evident etiology) demonstrated by cath, CT, or MR of:
Aneurysm, dilation
Narrowing or occlusion
Thickened arterial walls
Additionally, diagnosis requires one of the following five criteria:
Pulse deficit or claudication: lost or decreased or unequal peripheral artery pulses; focal muscle pain induced by activity
BP discrepancy: >10 mm Hg between any of the four limbs
Bruits: audible murmurs or palpable thrills over the larger arteries
Hypertension: SBP or DBP >95% for height
Acute phase reactant: ESR >20, CRP > normal
TA Epidemiology (Prevalence, Incidence)
The estimated incidence of TA is 2.6 per million, with higher prevalence in Southeast Asia, Central and South America, and Africa (3,33,34,35,136,137).
Although the disease has a strong predilection for young adult women, TA can occur in all age groups. While the true incidence in the pediatric population is not known, clearly it is a rarely reported disease, with the largest meta review of pediatric cases in the literature comprised of 241 cases (28,43,45,46,47,48,122,123). The largest US case series was comprised of only 21 patients (43,44,45,47,49). Seventy-five percent of pediatric patients are between 10 and 20 years of age, but infants as young as 1.5 months have been reported (7,27,28,31,32,60,62,63,64,65,66,67,68,73,105,107,118,124,125,138).
TA Etiology/Pathology
The cause of TA is unknown. An autoimmune T-cell–mediated process in those with a genetic susceptibility and influenced by environmental factors is suspected (69,81,109,122,123,126,127,136,137,139). TA has been reported in combination with a wide variety of other autoimmune diseases such as inflammatory bowel disease, lupus, and rheumatoid arthritis (33,69,70). In regions with endemic TB, positive PPDs have been reported in 29% to 90% of Takayasu patients (28,29,53,54,71,72,73,74,75,76,117,118,119,128,129,130). Whether this is coincidence, infectious, or immunopathogenic is not clear.
TA generates a panarteritis that progresses from adventitia to intima. Expression of heat shock protein-65 in response to an unknown stress appears to trigger the inflammatory cascade with a resultant vasculitic infiltrate of CD4 and CD lymphocytes, plasma cells, and macrophages (28,29,56,57,69,77,122,123,126,127,130). Giant cell granulomatous reaction and laminar necrosis ensue as well. Over time, there is a reactive fibrosis in the intima and neovascularization at the intimal–medial junction. Vascular pathology evolves to fibrosis and thickening in all layers, which effectively narrows or obliterates the vessel lumens (122,123,126,127,129,130). Skip lesions (more normal vessel in between segments of diseased vessel) are characteristic of TA.
Dilation and aneurysms develop when severe or rapid inflammation leads to destruction of the elastic media and smooth muscle cells with release of matrix metalloproteinases and other oxidants (28,140). Chronically, increased mural stress from hypertension or increased volume from aortic insufficiency (AI) may factor in the development of aneurysms later in the course of the disease as well (28,29,34,58,116,122,132,133,141).
TA Clinical Features
TA is classically described as having an active or systemic phase of vascular inflammation, followed by a chronic phase associated with arterial narrowing and occlusion. However, these phases may not be so readily distinguishable in an individual patient. The active phase may spontaneously remit after 3 months, or progress insidiously for months to years (127,129). As well, inflammatory and occlusive phases can coexist in different segments of the same vessel (28,142,143).
The extent and pattern of arterial involvement is the basis of several classification schemes for TA, including the following by Numano in 1996 (134,142).
Type I: Aortic arch and branches
Type II A: Ascending aorta, arch, and branches
Type II B: Ascending aorta, arch, descending thoracic aorta, and branches
Type III: Thoracic aorta and abdominal aorta
Type IV: Abdominal aorta
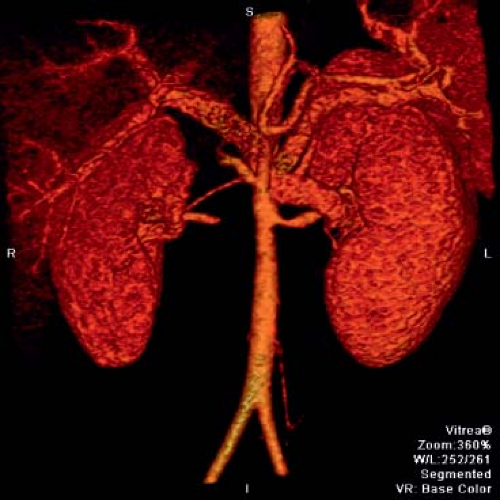
Figure 60.5 Takayasu arteritis patient with severe narrowing of the proximal left renal artery and obliteration of the right renal artery.
Type V: Combination of IIB and IV
C+: Coronary involvement
P+: Pulmonary involvement
Common sites of pediatric disease involvement include the thoracic and abdominal aorta (including renals) (Figs. 60.5 and 60.6) (69,72,109,120,122,135,144). Stenosis is the most common reported abnormality on imaging in the pediatric TA patient, followed by evidence for inflammation (29,31,63,65,136,137,145,146). The reported incidence of aneurysms in pediatrics varies widely, from 19% to 65% (147). The abdominal aorta is a frequent site of aneurysm development, but has been reported in the subclavians and thoracic aorta as well (81,139,145,146,148,149,150).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


