Genes associated with hypertrophic cardiomyopathy (HC) are not uniformly expressed in the atrial myocardium. Whether this may impact susceptibility to atrial fibrillation (AF) is unresolved. To analyze the prevalence and clinical correlates of AF in relation to genotype in a large HC cohort, prevalence and clinical profile of AF were assessed in 237 patients with HC, followed for 14 ± 10 years. Patients were divided into 3 genetic subgroups: (1) MYBPC3 (58%), (2) MYH7 (28%), and (3) “other genotypes” (14%; comprising TNNT2 , TNNI3 , TPM1 , MYL2 , complex genotypes, Z-line, and E-C coupling genes). Left atrial size was similar in the 3 subsets. AF occurred in 74 patients with HC (31%), with no difference among groups (31% in MYBPC3 , 37% in MYH7 and 18% in other genotypes, p = 0.15), paroxysmal/persistent AF (12%, 18%, and 12%, respectively; p = 0.53), paroxysmal/persistent evolved to permanent (12%, 12%, and 3%, p = 0.36) or permanent AF (7%, 7%, and 3%, p = 0.82). Age at AF onset was younger in the group with other genotypes (37 ± 10 years) compared to the first 2 groups (53 ± 14 and 51 ± 17, respectively; p = 0.05) because of early onset associated with complex genotypes and a specific JPH2 mutation associated with abnormal intracellular calcium handling. At multivariate analysis, independent predictors of AF were atrial diameter (p ≤0.05) and age at diagnosis (p = 0.09), but not genetic subtype (p = 0.35). In conclusion, in patients with HC, genetic testing cannot be used in clinical decision making with regard to management strategies for AF. Genotype is not predictive of onset or severity of AF, which appears rather driven by hemodynamic determinants of atrial dilatation. Exceptions are represented by rare genes suggesting specific molecular pathways for AF in genetic cardiomyopathies.
Hypertrophic cardiomyopathy (HC) is a complex genetically transmitted cardiac disease with diverse clinical course. The onset of atrial fibrillation (AF) is a frequent event in the course of HC, found in 20% to 25% of cases reported in the studies and related with adverse prognosis. The genesis of AF is generally attributed to hemodynamic factors such as diastolic dysfunction and outflow obstruction, leading to progressive left atrial enlargement. However, the genetic defects itself may directly increase susceptibility to AF, causing an intrinsic atrial myopathy in patients with HC. This hypothesis has been suggested by a large number of patients with HC showing an early onset of AF that cannot be explained exclusively with hemodynamic factors. In addition, in patients with HC, the efficacy of AF ablation is significantly lower compared with subjects without HC, suggesting a different AF substrate in patients with HC. We previously demonstrated that patients with HC with sarcomere myofilament mutations are more susceptible to AF compared to the myofilament negative. The genes most frequently responsible for HC (i.e., MYH7 encoding for β-myosin, MYBPC3 encoding for myosin-binding protein C, and genes encoding for thin filament proteins) are not equally and uniformly expressed in the atria and ventricles . Thus, it may be hypothesized that different genetic origins may directly impact individual patient predisposition to developing AF. In the present work, we took advantage of a large genotyped HC cohort to assess the prevalence, age at onset, and clinical correlates of AF based on molecular etiology of the disease.
Methods
The study was approved by the ethical committee of Careggi University Hospital (protocol number 2013/0035305). All patients enrolled gave individual informed consent. We assessed 237 unselected patients with HC, followed at the Reference Center for Cardiomyopathies of Careggi University Hospital in Florence, after the identification of a pathogenic/likely pathogenic disease–causing mutation by Sanger sequencing or next generation sequencing analysis. The diagnosis of HC was based on M-mode and 2-dimensional echocardiographic evidence of a hypertrophied left ventricle (LV; maximal wall thickness ≥15 mm or >95% confidence limits in relation to the body surface in children) in the absence of any other cardiac or systemic cause of LV hypertrophy. Based on genotype, patients were divided in 3 subgroups: myosin-binding protein C ( MYBPC3 ; 58%), β-myosin heavy chain ( MYH7 ; 28%), and other genotypes (14%), including genes of the thin filament ( TNNT2, TNNI3, TPM1 ) (5%), the thick filament MYL2 (4%), complex genotypes with >1 mutation (3%), Z-line (1%), and excitation–contraction (E-C) coupling genes (0.4%). Patients were followed up for 14 ± 10 years after initial diagnosis. Documentation of AF was based on electrocardiogram recordings obtained either after acute onset of symptoms or fortuitously during routine medical examination in asymptomatic patients. AF was defined as paroxysmal/persistent when it was either self-terminating or successfully cardioverted to sinus rhythm; paroxysmal/persistent and permanent AF was defined according to standand nomenclature. Specifically, paroxysmal AF was defined as AF that terminates spontaneously or with intervention within 7 days of onset, with episodes that may recur with variable frequency. Persistent AF was defined as continuous AF that is sustained >7 days. The term “permanent AF” is used when the patient and clinician make a joint decision to stop further attempts to restore and/or maintain sinus rhythm. In patients presenting with recent-onset AF, restoration of sinus rhythm was attempted by pharmacologic treatment or cardioversion after optimal anticoagulation with warfarin. Conversely, patients with permanent AF associated with severe left atrial dilatation were treated pharmacologically to achieve optimal ventricular rate control. Pharmacologic therapy during follow-up is reported in Table 1 .
| Overall n=237 | MYBCP n=137 (58%) | MHY7 n=67 (28%) | Other genotypes n=33 (14%) | p overall | |
|---|---|---|---|---|---|
| Initial evaluation | |||||
| Male | 150 (63%) | 92 (67%) | 38 (57%) | 20 (61%) | 0.35 |
| Age at enrollment (years) | 39±17 | 41±15 | 38±19 | 32±18 | 0.02 |
| Age at final evaluation (years) | 53±17 | 55±17 | 51±19 | 49±18 | 0.12 |
| Family history of HC | 114 (48%) | 72 (53%) | 24 (36%) | 18 (54%) | 0.06 |
| Family history of sudden cardiac death | 66 (28%) | 42 (31%) | 18 (27%) | 6 (18) | 0.36 |
| NYHA functional class at initial evaluation | |||||
| I | 124 (%) | 73 (53%) | 32 (48%) | 19 (58%) | 0.80 |
| II | 80 (34%) | 43 (31%) | 26 (39%) | 11 (33%) | |
| III | 24 (10%) | 13 (9%) | 8 (12%) | 3 (9%) | |
| Syncope | 45 (19%) | 22 (16%) | 14 (21%) | 9 (27%) | 0.29 |
| Prior cardiac arrest | 7 (3%) | 5 (4%) | 1 (1%) | 1 (3%) | 0.86 |
| Non-sustained ventricular tachycardia | 104 (44%) | 70 (51%) | 19 (28%) | 15 (45%) | 0.008 |
| Sustained ventricular tachycardia | 15 (6%) | 12 (9%) | 2 (3%) | 1 (3%) | 0.24 |
| Left atrium diameter (mm) | 43±8 | 43±9 | 43±8 | 43±7 | 0.97 |
| Left atrium volume (mL) | 85±50 | 83±35 | 89±75 | 86±31 | 0.71 |
| Maximum LV wall thickness (mm) | 23±6 | 23±6 | 23±7 | 23±7 | 0.99 |
| Maximal thickness site | |||||
| Septum | 220 (93%) | 126 (92%) | 64 (95%) | 30 (90%) | 0.90 |
| Apex | 7 (3%) | 4 (3%) | 2 (3%) | 1 (3%) | |
| Concentric | 9 (4%) | 6 (4%) | 1 (1%) | 2 (6%) | |
| LV End-diastolic diameter (mm) | 44±7 | 45±9 | 43±6 | 44±8 | 0.25 |
| LV End-systolic diameter (mm) | 26±8 | 27±8 | 25±7 | 26±8 | 0.22 |
| LV Ejectionfraction (%) | 64±10 | 63±10 | 66±10 | 64±10 | 0.13 |
| LV Ejectionfraction < 50% | 15 (6%) | 9 (7%) | 3 (4%) | 3 (9%) | 0.64 |
| LVOT Gradient (mmHg) | 21±24 | 19±24 | 25±24 | 19±28 | 0.24 |
| LVOT Obstruction (≥30mmHg) | 45 (19%) | 21 (15%) | 20 (30%) | 4 (12%) | 0.03 |
| LV Filling pattern | |||||
| Normal | 56 (28%) | 33 (30%) | 19(34%) | 4 (13%) | 0.4 |
| Impaired relaxation | 89 (45%) | 47 (42%) | 24(43%) | 18(58%) | |
| Pseudonormal | 36 (18%) | 21 (19%) | 9 (16%) | 6 (19%) | |
| Restrictive | 15 (7%) | 10 (9%) | 3 (5%) | 2 (6%) | |
| Triphasic | 2 (1%) | 0 (0%) | 1 (2%) | 1 (3%) | |
| Final evaluation | |||||
| Age at final evaluation (years) | 53±17 | 55±17 | 51±19 | 49±18 | 0.12 |
| NYHA functional class at final evaluation | |||||
| I | 107 (45%) | 65 (47%) | 29 (43%) | 13 (39%) | 0.68 |
| II | 96 (41%) | 51 (37%) | 31 (46%) | 14 (42%) | |
| III | 28 (12%) | 18 (13%) | 5 (7%) | 5 (15%) | |
| IV | 3 (1%) | 2 (1%) | 1 (1%) | 0 (0%) | |
| Left atrium diameter (mm) | 45±9 | 46±10 | 46±9 | 41±6 | 0.86 |
| Left atrium volume (cc) | 97±49 | 101±52 | 95±50 | 89±31 | 0.40 |
| Maximum LV wall thickness (mm) | 22±6 | 22±5 | 22±6 | 21±6 | 0.62 |
| LV End-diastolic diameter (mm) | 47±7 | 47±8 | 45±6 | 47±5 | 0.15 |
| LV End-systolic diameter (mm) | 29±8 | 29±9 | 27±7 | 30±8 | 0.16 |
| LV Ejection fraction (%) | 62±12 | 62±13 | 65±10 | 59±14 | 0.06 |
| LV Ejection fraction < 50% | 28 (12%) | 19 (14%) | 4 (6%) | 6 (18%) | 0.12 |
| LVOT Gradient (mmHg) | 12±12 | 11±12 | 15±14 | 9±5 | 0.03 |
| LVOT Obstruction (≥30mmHg) | 44 (19%) | 20 (15%) | 22 (33%) | 2 (6%) | 0.001 |
| LV Filling pattern | |||||
| Normal | 24 (10%) | 16 (12%) | 7 (10%) | 1 (3%) | 0.03 |
| Impaired relaxation | 40 (17%) | 19 (14%) | 15 (22%) | 6 (18%) | |
| Pseudonormal | 89 (37%) | 46 (34%) | 32 (48%) | 11 (33%) | |
| Restrictive | 42 (18%) | 29 (21%) | 8 (12%) | 5 (15%) | |
| Triphasic | 8 (3%) | 3 (2%) | 1 (1%) | 4 (12%) | |
| Interventions during follow-up | |||||
| Myectomy | 35 (15%) | 18 (13%) | 13 (19%) | 4 (12%) | 0.46 |
| Alcohol ablation | 26 (11%) | 16 (12%) | 7 (10%) | 3 (9%) | 0.9 |
| Surgical teraphy-cardiac valve | 19 (8%) | 11 (8%) | 8 (12%) | 0 (0%) | 0.10 |
| Other cardiac surgery teraphy | 6 (3%) | 4 (3%) | 2 (3%) | 0 (0%) | 0.86 |
| Implantablecardioverter-defibrillation | |||||
| ICD | 58 (24%) | 30 (22%) | 15 (22%) | 13 (39%) | 0.02 |
| PM | 9 (4%) | 5 (4%) | 4 (6%) | 0 (0%) | |
| CRT-D | 9 (4%) | 6 (4%) | 0 (0%) | 3 (9%) | |
| General outcome | |||||
| Follow-up (years) | 14±11 | 14±10 | 13±11 | 17±12 | 0.20 |
| Unfavorable outcome | 86 (36%) | 49 (36%) | 21 (31%) | 16 (48%) | 0.24 |
| Progression to NYHA class III or IV | 23 (10%) | 15 (11%) | 5 (7%) | 3 (9%) | 0.82 |
| Death for cardiovascular causes: | |||||
| Heart failure | 6 (19%) | 4 (31%) | 2 (25%) | 0 (0%) | 1.0 |
| Sudden Death or appropriate ICD discharges | 7 (23%) | 5 (38%) | 2 (25%) | 0 (0%) | 1.0 |
| Hospitalizations for heart failure | 24 (10%) | 16 (12%) | 5 (7%) | 3 (9%) | 0.72 |
| Non-fatal stroke | 15 (6%) | 10 (7%) | 5 (7%) | 0 (0%) | 0.31 |
| Therapy during follow-up | |||||
| Amiodarone Initial evaluation | 33 (14%) | 16 (12%) | 11 (16%) | 6 (18%) | 0.05 |
| Final evaluation | 44 (19%) | 34 (25%) | 7 (10%) | 3 (9%) | |
| Beta Blockers Initial evaluation | 34 (14%) | 25 (18%) | 7 (10%) | 2 (6%) | 0.03 |
| Final evaluation | 158 (67%) | 83 (61%) | 53 (79%) | 22 (67%) | |
| Ca antagonists Initial evaluation | 44 (19%) | 31 (23%) | 8 (12%) | 5 (15%) | 0.23 |
| Final evaluation | 36 (15%) | 19 (14%) | 11 (16%) | 6 (18%) | |
| Disopyramide Initial evaluation | 19 (8%) | 9 (7%) | 9 (13%) | 1 (3%) | 0.08 |
| Final evaluation | 7 (3%) | 3 (2%) | 4 (6%) | 0 (0%) | |
| Other antiarrithmic Initial evaluation | 11 (5%) | 10 (7%) | 1 (1%) | 0 (0%) | 0.007 |
| Final evaluation | 14 (6%) | 11 (8%) | 2 (3%) | 1 (1%) | |
| ACE-I Initial evaluation | 24 (10%) | 17 (12%) | 2 (3%) | 5 (15%) | 0.08 |
| Final evaluation | 34 (14%) | 18 (13%) | 9 (13%) | 7 (21%) | |
| ARB Initial evaluation | 7 (3%) | 4 (3%) | 2 (3%) | 1 (3%) | 0.65 |
| Final evaluation | 23 (10%) | 13 (9%) | 5 (7%) | 5 (15%) | |
| Statins Initial evaluation | 6 (2%) | 5 (4%) | 1 (1%) | 0 (0%) | 0.22 |
| Final evaluation | 31 (13%) | 20 (15%) | 6 (9%) | 5 (15%) | |
| Warfarin Initial evaluation | 13 (5%) | 6 (4%) | 6 (9%) | 1 (3%) | 0.18 |
| Final evaluation | 57 (24%) | 38 (28%) | 15 (22%) | 4 (12%) | |
| Aspirin Initial evaluation | 22 (9%) | 16 (12%) | 5 (7%) | 1 (3%) | 0.06 |
| Final evaluation | 39 (16%) | 24 (17%) | 11 (16%) | 4 (12%) | |
After genetic counseling and informed consent, genomic DNA was extracted from peripheral blood by automated standard protocols. Patients were screened for mutations in 8 protein-coding exons and splice sites of HC-causing genes, including MYBPC3 , thick filament proteins ( MYH7 and the regulatory and essential light chains), and thin filament proteins (troponin T, troponin I, alpha-tropomyosin, and alpha-actin) by conventional Sanger sequencing. Furthermore, in selected patients in whom the initial panel was negative, additional genes were analyzed including ACTN2, CSPR3, TCAP, VCL, MYOZ2, ZASP and JPH2 by targeted massively parallel sequencing. To identify clinically relevant variants, data were analyzed based on minor allele frequency (≤0.01) using dbSNP Release 138 ( http://ncbi.nlm.nih.gov/projects/SNP ), Exome Sequencing Project ( http://evs.gs.washington.edu/EVS/ ), 1,000 genomes project ( http://www.1000genomes.org ), and Exome Aggregation Consortium ( http://exac.broadinstitute.org/ ). Subsequently, the variants were analyzed according to the location (coding, 5′ or 3′ junctions), the effect on protein, the evolutionary conservation of the affected nucleotide (PhyloP score) and pathogenic potential by in silico predictive algorithms such as MutationTaster, Sorting Intolerant From Tolerant, and PolyPhen-2. The Human Gene Mutation Database Professional was used to identify previously reported variants. Sanger sequencing was also used to confirm the presence of the variants found by next generation sequencing and to perform cosegregation analysis. In addition to classify variants, we used the Alamut Visual software, version 2.6 (Interactive Biosoftwares, Rouen, France). By applying the previously mentioned rules, both the variants found by conventional Sanger and that by next generation sequencing were classified in 4 main classes such as known pathogenic, likely (probably) pathogenic, variants of unknown significance, and benign variants. In this study, only pathogenic and likely pathogenic variants were included.
Unpaired Student t test or 1-way analysis of variance was used for the comparison of normally distributed data. The chi-square or Fisher’s exact test were used to compare noncontinuous variables expressed as proportions. Relations between independent variables were assessed with Spearman bivariate correlation analysis. Hazard ratios (HRs) and 95% CIs (CIs) were calculated using univariate logistic regression analysis. All calculations were performed using SPSS software.
Results
Mean age at diagnosis of the 237 patients with HC was 39 ± 17 years; 37% (n = 87) were women ( Table 1 ). Overall, 48% of patients (n = 114) had a positive family history of HC and 28% of sudden cardiac death. At initial evaluation, maximal LV wall thickness was 23 ± 6 mm and was mainly localized at the level of the interventricular septum (93%). In 19% of patients, we observed LV outflow tract obstruction at rest (≥30 mm Hg). The pattern of diastolic filling was normal in 56 patients (28%), compatible with impaired relaxation in 89 (45%), pseudonormal in 36 (18%), and restrictive in 15 (7%). Average left atrium (LA) diameter was 43 ± 8 mm and exceeded 50 mm in 34 patients; average LA volume was 85 ± 50 cc.
Of the 237 patients, 7 patients (3%) had a history of resuscitated cardiac arrest and 15 (6%) of sustained ventricular tachycardia. A family history of sudden cardiac death was present in 66 patients (28%) and a history of syncope in 45 patients (19%). In addition, 104 patients (44%) experienced repeated episodes of nonsustained ventricular tachycardia on Holter electrocardiogram tracing; massive LV hypertrophy ≥30 mm was present in 24 patients (10%). An internal cardioverter–defibrillator was implanted in 58 patients (24%) as primary or secondary prevention. In addition, 35 patients (15%) underwent surgical septal myectomy and 26 (11%) alcohol septal ablation for symptoms associated to outflow tract obstruction refractory to drug therapy.
A total of 266 mutations were found in the 237 patients, including 148 missense, 66 splicing mutations, 44 frame-shift, and 8 nonsense mutations. The gene most frequently associated with HC was MYBPC3 , in 137 patients (58%) and, in them, the most frequently observed was MYBPC3-E258K , because of a founder effect known in the HC population of the Florentine area. MYBPC3-E258K mutation was present in 54 patients (39% of the total number of MYBPC3 mutations carriers). Furthermore, 76 patients (32%) had mutations in genes encoding for myosin heavy or light chains (67 in MYH7 and 9 in MYL2 ), and 12 (5%) in thin filament genes (7 in TNNT2 , 1 in TPM1 , and 4 in TNNI3 ). A complex genotype was present in 7 patients in which 2 mutations were found in 2 different genes. There is also a case of triple mutation, which shows mutations in the genes TNNI3, MYBPC3 , andMYH7 . In 4 genotype negative patients, we peformed further analysis or rare HC-associated genes, that is, ACTN2, CSPR3, ZASP, TCAP, VCL, MYOZ2 , andJPH2 . Two of the 4 were positive for ZASP mutations and one had the E169K JPH2 mutation, described in more detail in the following section. Type 2 junctophillin ( JPH2 ) is a membrane protein involved in the E-C coupling mechanism and located in correspondence of the Z-lines. During a mean follow-up of 14 ± 10 years from initial diagnosis, AF was documented in 74 patients, with a prevalence of 31%. The first manifestation of AF was paroxysmal/persistent in 58 patients (with subsequent evolution to permanent AF in 25) and persistent/permanent in 16 patients. The age at onset of AF was 51 ± 15 years; in 22% of patients, there was early onset <40 years. The paroxysmal/persistent form started at a slightly (but not significantly) younger age than permanent AF (50 ± 15 vs 56 ± 14 years, respectively, p = 0.8). In the 25 patients with both manifestations, the median time from paroxysmal/persistent to permanent AF was 6 years.
In most of the 74 patients (n = 65; 88%), the onset of AF was associated with new symptoms or worsening of existing symptoms, such as dyspnea in 53 patients (72%), palpitations in 40 patients (54%), angina in 11 patients (15%). Occasionally, the acute phase of paroxysmal/persistent AF associated with a rapid ventricular response elicited pulmonary edema (n = 3 or 1%) and presyncope (n = 7 or 9%). Only in a minority of patients (n = 9 or 12%), AF was documented accidentally during routine evaluations, in the absence of reported symptoms. During paroxysms of AF, 10% of patients were in New York Heart Association (NYHA) class I, 52% in class II, and 33% in class III/IV. Of those patients with permanent AF, 10% were in NYHA class I, 41% in class II, and 49% in class III/IV. Cardioembolic events occurred in 8 of the 74 patients with AF (11%), including 4 on warfarin. Overall, 86 of the 237 patients (36%) had an unfavorable outcome, including heart failure death, sudden death, or appropriate internal cardioverter–defibrillator interventions or progression to severe functional limitation (NYHA class III/IV; Table 1 ).
Most patients (93%) received drug treatment for HC including β blockers, calcium channel blockers, amiodarone, and disopyramide ( Table 1 ). Of the 74 patients with AF, 44 received antiarrhythmic therapy with amiodarone (59%), 65 with β blockers (88%), and 8 with disopyramide (11%, in the presence of outflow obstruction). Furthermore, 57 patients (24%) were on oral anticoagulation with warfarin; the remaining 17 patients were on antiplatelet agents (including 11 who declined warfarin). Eighteen patients (24%) underwent radiofrequency catheter ablation for symptomatic AF refractory to drug therapy. Of these, 11 remained in stable sinus rhythm (although all but one retained antiarrhythmic therapy); in the remaining 6, AF recurred and became permanent.
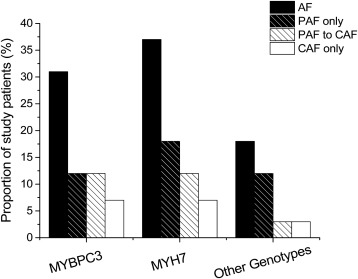
For the purpose of assessing the predisposition to AF, the study population was divided into 3 groups based on genotype: MYBPC3 (58%), MYH7 (28%), and other genotypes (14%, including mutations in other genes and complex genotypes). Of note, the distribution of mutations reflects that of the general HC population in our Florence cohort (53% MYBPC3 , 32% MYH7 , 11% other genotypes), previously described by Olivotto et al. The 3 groups were comparable in terms of prevalence of AF (31%, 37%, and 18%, respectively, p = 0.15), whether paroxysmal/persistent (12%, 18%, and 12%), paroxysmal/persistent evolved to permanent (12%, 12%, and 3%) or permanent AF (7%, 7%, and 3%; p >0.1 for all comparisons), Figure 1 and Table 2 . Age at the time of first documentation of AF was similar in MYBPC3 and MYH7 (53 ± 14 and 51 ± 17, respectively), whereas the onset was earlier in the other genotypes group at a mean age of 37 ± 10 years (p <0.05 vs the other 2 groups; Figure 2 ), because of an unusually early onset of paroxysmal/persistent AF. Conversely, we found no difference in age at onset of permanent AF.