Impact of Cardiac Disease on Other Organ Systems
Jack Rychik
Structural malformations of the heart makeup approximately 1% of all forms of birth defects in humans. When present, congenital heart disease (CHD) may also be associated with changes in organ systems outside of the heart and blood vessels. Considering the role of the heart as the engine of circulatory propulsion, cardiac structural aberrations may impact capacity for blood flow delivery, with important downstream consequences affecting end-organ functionality. Organs not receiving adequate blood flow with decreased oxygen delivery may malfunction, or develop poorly, leading to long-term complications. There can also be consequences following treatment of heart disease in the developing, immature human infant or child while organs are in a vulnerable, at risk state. For most forms of CHD, there is no absolute “cure,” no capacity to fully replicate a completely normal cardiovascular system after intervention such as surgery. Even in cases that are deemed successfully treated by current standards, residual hemodynamic disturbances may persist which can exert either acute, overt changes or indolent, subclinical alterations in end-organ performance. Organ system malfunction may be evident and manifest anywhere from early in infancy to later in life in the adult survivor with CHD.
In this chapter, we will discuss the impact of CHD on organ systems outside of the heart. Details of two commonly affected areas—the neurologic (Chapter 74) and hematologic systems (Chapter 75)—and their relationship to CHD, are reviewed elsewhere in other chapters and will not be addressed here.
How and Why are other Organ Systems Affected in CHD?
A number of potential mechanistic models and conceptual frameworks explain why extracardiac organ systems may be negatively affected when there is CHD. First, organs themselves may be primarily malformed in association with CHD (1). Additional extracardiac findings are often identified upon comprehensive evaluation when CHD is present, suggesting the importance of screening (2). In a large series from France of over 4,000 cases of CHD, 26% had associated major anomalies. Chromosomal or genetic anomalies influence multiple developing cell lines and often result in abnormalities of both the cardiac and noncardiac systems. Anomalies associated with CHD can be placed into a recognizable syndrome or pattern in approximately 12% of cases with CHD (3). In another large series of 3,751 births of infants with CHD in Alberta, Canada from 1995 to 2002, 75% had isolated heart anomalies, 10% had a chromosomal etiology, and 9% had multiple major extracardiac anomalies. The most commonly associated major noncardiac anomalies were musculoskeletal (24%), anomalies of the urinary tract (14%), gastrointestinal system (11%), and central nervous system (11%) (4).
As an example of a chromosomal etiology and model for CHD and extracardiac anomalies, a newborn with trisomy 21 may exhibit an atrioventricular septal defect, as well as duodenal atresia, an abnormality of the gastrointestinal system, both of which demand treatment early in life (5). Such a child would also be at risk for a multitude of additional noncardiac consequences, including dysmorphology, neurodevelopmental delay, musculoskeletal abnormalities, and a host of other organ system abnormalities. These findings have their origin in the nondisjunction of chromosome 21, resulting in the constellation of findings recognized as Down syndrome.
Patterns of phenotypic findings may recur in various patients suggesting an association, although a genetic explanation may not be all that clear. For some patients, constellations of cardiac and extracardiac findings fall into a “syndrome,” with as of yet ill-defined specific chromosomal or genetic origins. One such example would be the “VACTERL association,” a condition that includes vertebral anomalies, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities (6). Patients with at least three of these characteristic features are considered to carry the diagnosis. Defining this condition through the presence of specific individual findings indicating a “phenotype” highlights the arbitrary nature of the threshold for diagnosis. What we currently call “VACTERL association” is a term for a broad categorical diagnosis, likely including a number of different specific conditions with variable genetic, or environmental origins. The common thread is an early disruption in development along multiple cell lines leading to changes in both cardiac and extracardiac systems resulting in the phenotype described.
Heterotaxy syndrome is another example of a more pervasive multiorgan system abnormality with both cardiac and extracardiac manifestations. A defect in lateralization commonly affects cardiac structure in a host of manners, and is associated with abdominal organ abnormalities affecting the spleen, liver, and small bowel. Ciliary dyskinesia is a common finding in heterotaxy syndrome and possibly points to the origins of the syndrome (7). Ciliary function is an important contributor to organ positioning during early development, and early primary ciliary dysfunction related to mutations in dynein leads to abnormal organ positioning (8). Abnormal capacity to localize early primitive structures alters the potential for normal right–left symmetry, influencing cardiac anatomical development such as looping and chamber positioning. The residual of early gestational ciliary dysfunction can manifest later in life as clinical findings due to ciliary malfunction such as recurrent sinus infections, respiratory infections due to impaired mucociliary clearance, or possible male reproductive infertility due to sperm tail dysfunction (9) (Fig. 76.1). Respiratory consequences of ciliary dyskinesia may also negatively impact recovery after surgical intervention in the heterotaxy population (10).
Subtle associations may exist between CHD and extracardiac organ malformations. In a sense, all forms of CHD are by definition congenital malformations influenced by an abnormality or a misread of the “blueprint” signaling normal cardiovascular development. While the cardiac anomaly may be immediately evident, ostensibly transparent and clinically important, other organ systems may also be affected with a structural or functional difference, but perhaps in a nonclinically apparent and subtle manner. Inconspicuous extracardiac organ system differences may only become
apparent through greater detailed evaluation early in life. Sometimes, such differences remain clinically silent for the duration of a lifespan, or they may cross the threshold of clinical manifestation only after growth and further development have taken place, or simply through the passage of time.
apparent through greater detailed evaluation early in life. Sometimes, such differences remain clinically silent for the duration of a lifespan, or they may cross the threshold of clinical manifestation only after growth and further development have taken place, or simply through the passage of time.
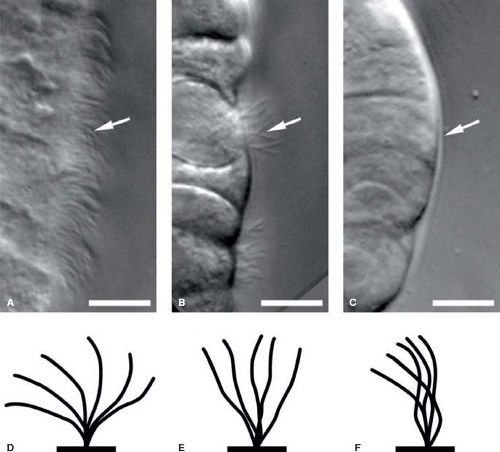 Figure 76.1 A–C: Nasal epithelia from heterotaxy patients with ciliary dyskinesia. D–F: Diagrammatic representation of nasal cilia. A: Abundant cilia (arrow) exhibiting normal ciliary motion. B: Paucity of cilia (arrow). C: No cilia found in nasal epithelium (arrow). D: Diagram of healthy architecture of cilia. E: Diagram of limited stroke capacity with minimal ciliary bend. F: Diagram of very abnormal ciliary bending. (From Nakhleh N, Francis R, Giese RA, et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125:2232–2242.) |
One such example is tetralogy of Fallot. Approximately 20% of affected individuals have a defined, currently known “blueprint” origin, most commonly either 22q11 microdeletion (11), trisomy 21 (12), a mutation in the notch pathway ligand Jagged 1 (13), or NKX2.5 mutation (14), or other rare etiology. Conversely, 80% do not have a clear chromosomal, genetic association. Clinical features seen in 22q11 deletion, the DiGeorge syndrome, such as learning difficulties may not be evident until a child is placed in the school environment and challenged to perform. Patients with Jagged 1 mutation, the Alagille syndrome, may have subclinical or very subtle characteristics such as mild facial dysmorphology, or manifest very mild liver abnormalities, evident only in adolescence or adulthood. Such patients may have tetralogy of Fallot diagnosed and treated early in life, but yet have differences in other organ systems that without detailed initial investigation or the passage of time, may go undetected. It is safe to say that patients with CHD who may have well-functioning organs while young, are at risk for manifesting organ system complications later in life.
Another possible mechanistic model for extracardiac organ system abnormality may be due to an acquired change during fetal life. Primary malformations of the heart alter blood flow patterns that may influence oxygen delivery leading to secondary changes in various organ systems. One such example which illustrates a possible model is the condition of transposition of the great arteries. In transposition of the great arteries, right-to-left streaming of relatively highly oxygenated blood from the ductus venosus across the atrial septum leads to relatively highly oxygenated blood delivery to the left ventricle, pulmonary artery, and ductus arteriosus. Increased pulmonary arterial oxygen tension in transposition of the great arteries can lead to alterations in pulmonary vascular development. Conversely, relatively poorly oxygenated blood is directed toward the right ventricle and aorta. Hence, in this anatomical configuration the coronary and cerebral circulations receive a somewhat lower
blood oxygen content than normal, and the abdominal organs, perfused by the postductal ejection of oxygenated blood from the left ventricle, receive a higher blood oxygen content than normal. Alterations in brain structure and functional neurocognitive deficits have been identified in children with transposition of the great arteries. Whether alterations in flow and oxygen delivery are contributors to these findings is somewhat inconclusive at this point in time. In utero hypoxia can lead to epigenetic programming with important influences on organ ontogeny, structure, and function (15). Interestingly, of all the various forms of complex CHD in the neonate, transposition of the great arteries has been the least associated with any genetic or syndromic cause, and is most commonly seen in an isolated manner with little extracardiac organ malformation (16). Transposition of the great arteries may therefore be an ideal model to study as an example of acquired end-organ changes related to in utero alterations in oxygen delivery and not due to the presence of a primary syndrome affecting organ dysfunction.
blood oxygen content than normal, and the abdominal organs, perfused by the postductal ejection of oxygenated blood from the left ventricle, receive a higher blood oxygen content than normal. Alterations in brain structure and functional neurocognitive deficits have been identified in children with transposition of the great arteries. Whether alterations in flow and oxygen delivery are contributors to these findings is somewhat inconclusive at this point in time. In utero hypoxia can lead to epigenetic programming with important influences on organ ontogeny, structure, and function (15). Interestingly, of all the various forms of complex CHD in the neonate, transposition of the great arteries has been the least associated with any genetic or syndromic cause, and is most commonly seen in an isolated manner with little extracardiac organ malformation (16). Transposition of the great arteries may therefore be an ideal model to study as an example of acquired end-organ changes related to in utero alterations in oxygen delivery and not due to the presence of a primary syndrome affecting organ dysfunction.
Alterations in fetal blood flow patterns in CHD may set subtle changes into motion that are not evident early in life, but later may influence organ functionality. Barker proposed the notion that the fetal environment is dictated by maternal nutritional state (17) and that other extrinsic factors such as maternal stress (18) may influence life-long health status through “fetal programming” (19). One might speculate that changes in blood volume and oxygen delivery in utero due to CHD may contribute to “programmed” alterations in organ function, which may manifest later in life as well. Longitudinal studies of patients with prenatally diagnosed CHD are necessary in order to better answer these questions.
In addition to the association of CHD with primary extracardiac abnormalities, several unique conditions exist in which specific organ systems may be affected by CHD or its treatment.
Impact of CHD on the Renal System
The kidney is a highly vascularized structure, with renal function very much dependent upon blood perfusion and oxygen delivery. At birth, neonatal serum creatinine levels reflect maternal renal function, but within 24 to 48 hours begin to reflect infant renal function. A variety of forms of CHD may result in renal hypoperfusion and impaired renal function shortly after birth. In left-sided heart disease with ductal dependence for systemic perfusion such as in the hypoplastic left heart syndrome (HLHS) or coarctation of the aorta, a closing ductus arteriosus will impair the capacity for systemic perfusion leading to significantly diminished blood flow to the kidneys. Prenatal identification or early postnatal detection with commencement of prostaglandin infusion will preserve ductal patency, however, retrograde diastolic flow through the ductus arteriosus may “steal” blood flow away from the renal arteries leading to hypoperfusion, hence creating the potential for acute renal dysfunction.
With increasing frequency of fetal diagnosis and improved neonatal management strategies for maintaining systemic perfusion, the frequency of preoperative neonatal renal dysfunction is now relatively low. In a large multicenter study, prenatal diagnosis of single ventricle type of CHD was associated with a significantly lower rate of preoperative renal insufficiency in comparison to postnatal diagnosis (20). Abundant data on the frequency of acute renal dysfunction in the neonate with CHD are limited, partly due to lack of uniform definition. The Multi-Societal Database Committee for Pediatric and Congenital Heart Disease defines acute renal dysfunction as “new onset oliguria with sustained urine output less than 0.5 cc/kg/h for 24 hours and/or a rise in creatinine greater than 1.5 times upper limits of normal for age (or twice the most recent preoperative/preprocedural values if these are available), with eventual recovery of renal function without needing dialysis (including peritoneal dialysis and/or hemodialysis) or hemofiltration” (21). The definition of acute renal failure is the same but
“…with eventual need for dialysis (including peritoneal dialysis and/or hemodialysis) or hemofiltration.” Thus, renal dysfunction refers to transient azotemia and transient oliguria not requiring dialysis or hemofiltration, while renal failure requires temporary hemofiltration, temporary dialysis, or permanent dialysis.
Besides acute hemodynamic compromise and hypoperfusion due to hypotension either at presentation or perioperatively, renal function remains relatively well preserved in most patients with CHD, at least through the first decades of life. This may be related to the deep reserve of functionality and robustness built into the design of the human kidney, in which a substantial decrease in glomerular filtration is necessary before changes can be seen in fluid and electrolyte homeostasis. Nevertheless, from the perspective of kidney disease, CHD is the most common cause of acute renal failure in infancy. In general, those with good repair of CHD and without significant residual cyanosis typically have renal function quite comparable to normal (22).
Surgery for CHD may create a physiologic stress, leading to decreased renal blood flow, shift of renal blood flow from the renal cortex to the medulla, decrease in glomerular filtration rate (GFR), and release of vasopressin with fluid accumulation. Duration of cardiopulmonary bypass and hypothermic circulatory arrest are risk factors for renal dysfunction. Postoperative medications may be nephrotoxic, and in combination with fragile blood supply, may exert deleterious effects (23). Renal oximetry assessment through regional near-infrared spectroscopy has shown a relationship between low renal oximetry readings and peak postoperative serum creatinine levels (24). Renal failure defined as a serum creatinine >1.5 mg/dL, tripling of creatinine over <7 days, or need for dialysis was noted in 46/549 infants who underwent neonatal surgery for HLHS, arguably one of the most physiologically stressful operations for CHD (25).
Cyanosis exacerbates the potential for renal insufficiency in CHD. Cardiopulmonary bypass in patients with chronic cyanosis substantially increases the risk for postoperative renal dysfunction (26). Cyanosis leads to polycythemia with resulting blood hyperviscosity and may contribute to a diminished filtration fraction at the glomerular level (27). Histopathologic changes of the kidney have been noted on autopsy of children with cyanotic CHD. In one study, at a mean age of 4.6 years, renal histopathologic changes observed included glomerulomegaly, glomerulosclerosis, periglomerular fibrosis, hyperplastic arteriolosclerosis, and interstitial fibrosis. Interestingly, the objectively measured parameters were higher in patients with CHD having decreased pulmonary arterial blood flow than those having normal to increased pulmonary arterial blood flow (28).
Renal dysfunction is often appreciated in adult survivors of CHD. In one large series of over 1100 patients with CHD at an average age of 36 years, GFR was calculated and graded as normal, mild, moderate, or severe renal dysfunction based on degree of reduction in GFR (29). Renal dysfunction was mild in 41% of patients and moderate or severe in 9%. Decreased GFR was common among patients with Eisenmenger syndrome (72%). Overall mortality was threefold higher in those with moderate or severe reduction in GFR. Even in patients with tetralogy of Fallot, for which long-term survival following repair is excellent, over 50% exhibit evidence for reduced GFR and are therefore at risk for further kidney injury if exposed to an additional risk factor such as onset of type 2 diabetes or chronic use of diuretics (30).
Impact of CHD on the Gastrointestinal System
Primary congenital anomalies of the gastrointestinal system may occur concomitantly with CHD. Anomalies such as esophageal fistula, small bowel atresia or stenosis, gut malrotation, or biliary atresia have their own well-defined natural histories and established therapeutic strategies. Management protocols as well as prognosis
may however be affected by the presence of complex CHD. Healing following surgery for extracardiac conditions can be influenced by changes in regional perfusion and oxygen delivery. Dysphagia, feeding difficulties, and gastroesophageal reflux are common challenges in the infant with CHD, in particular following surgery, with etiologies that are complex and multifactorial. In general, the presence of congenital anomalies of the gastrointestinal tract may alter outcome for CHD by prolonging the recuperative period, as nutrition is key to a rapid recovery after surgery for CHD.
may however be affected by the presence of complex CHD. Healing following surgery for extracardiac conditions can be influenced by changes in regional perfusion and oxygen delivery. Dysphagia, feeding difficulties, and gastroesophageal reflux are common challenges in the infant with CHD, in particular following surgery, with etiologies that are complex and multifactorial. In general, the presence of congenital anomalies of the gastrointestinal tract may alter outcome for CHD by prolonging the recuperative period, as nutrition is key to a rapid recovery after surgery for CHD.
Altered blood flow and decreased oxygen delivery to the gastrointestinal system may occur secondary to CHD either before or after surgery. Gut ischemia can lead to frank infarction of tissue. Alterations in gut circulation in the at-risk, immature newborn intestine can lead to the condition of necrotizing enterocolitis (NEC). The Multi-Societal Database Committee for Pediatric and Congenital Heart Disease defines NEC as “an acute reduction in the supply of oxygenated blood to the small intestine or large intestine, typically resulting in acidosis, abdominal distention, pneumatosis, and/or intestinal perforation, that prompts initiation of antibiotics or exploratory laparotomy” as treatment (31). If NEC is present with acute evidence of frank ischemia and a break in intestinal integrity such as in association with radiographic evidence of intestinal pneumatosis, urgent surgery may be necessary to remove infarcted bowel tissue in order to avoid hemodynamic decompensation. The finding of NEC treated medically can lead to scarring after healing of affected bowel regions, with later development of stricture and possible need for further surgery. A staging system for NEC based on clinical, radiographic, and gastrointestinal criteria has been developed (32) (Table 76.1).
While a common problem in the premature infant, NEC can be seen as a complication in the full-term infant with CHD. In cardiac anomalies for which patency of the ductus arteriosus is necessary, potential steal of blood from the systemic to the pulmonic circulation may occur, raising concerns about the risk of gut ischemia and development of NEC. Feeding such infants with placement of nutritional material in the gut may on theoretical grounds, further increase the risk of local mesenteric ischemia. The proposed mechanism is one of mismatch between blood flow and oxygen delivery and the demand for perfusion, which may be increased during feeding. This rationale has been the focus of debate for many years as to the wisdom of feeding neonates with critical ductal dependent CHD. More recently, several centers have moved toward administering some degree of feeds in these neonates before surgery. Small amounts of preoperative “trophic” feeds may be beneficial, with some in fact arguing that it may reduce the incidence of NEC (33) or lead to earlier postoperative feeding tolerance in neonates with HLHS (34). In an analysis of over 6,700 infants with ductal-dependent CHD receiving prostaglandin E1 infusion, NEC occurred with an incidence of only 0.3%, and half of the patients who experienced it where premature at less 37 weeks’ gestation (35). Enteral feeding was not associated with increased odds of NEC. A risk factor associated with NEC was the diagnosis of single ventricle type of CHD, although the overall risk in this series remained low at incidence of 0.5%.
TABLE 76.1 Modified Bell’s Staging Criteria for Necrotizing Enterocolitis | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Can we get a better sense of the pathophysiologic origins of NEC in CHD? Focusing in on those with CHD in which NEC is most common may offer some insight. Conditions such as HLHS, truncus arteriosus, and aortopulmonary window are at increased risk for NEC—all lesions in which there is potential for torrential diastolic runoff and steal into the pulmonary circulation, placing the gut at risk for hypoperfusion (36).
While the incidence of preoperative NEC—if one looks at all forms of CHD—is generally relatively low, rates of postoperative
NEC in HLHS after Norwood operation are quite high. In one series, 18% of neonates after stage 1 Norwood palliation had evidence of NEC (37), while in a multicenter study NEC defined narrowly as the presence of pneumatosis intestinalis or free air on radiography was noted in 2.5% (25). Poor mesenteric perfusion after Norwood operation may be related to poor right ventricular systolic performance (pump failure), diastolic steal through a modified Blalock–Thomas–Taussig shunt, or perhaps a combination of the two affecting the mesenteric circulation. Interestingly, neonates who undergo hybrid procedure approach to first-stage reconstruction for HLHS, in which there is no diastolic runoff through a shunt, continue to have a relatively high rate of NEC, approximately 12% in one series (38). Reconstruction of the aorta results in a dramatic difference in flow kinetics in the descending aorta, which may modify the flow characteristics of delivery of blood to the mesenteric circulation. Echocardiographic evaluation of the descending aorta demonstrates a lower aortic pulsatility in association with NEC, suggesting the possibility of an inherent abnormality of the systemic vasculature, which may be a factor in predisposing to NEC in the postoperative HLHS population (39).
NEC in HLHS after Norwood operation are quite high. In one series, 18% of neonates after stage 1 Norwood palliation had evidence of NEC (37), while in a multicenter study NEC defined narrowly as the presence of pneumatosis intestinalis or free air on radiography was noted in 2.5% (25). Poor mesenteric perfusion after Norwood operation may be related to poor right ventricular systolic performance (pump failure), diastolic steal through a modified Blalock–Thomas–Taussig shunt, or perhaps a combination of the two affecting the mesenteric circulation. Interestingly, neonates who undergo hybrid procedure approach to first-stage reconstruction for HLHS, in which there is no diastolic runoff through a shunt, continue to have a relatively high rate of NEC, approximately 12% in one series (38). Reconstruction of the aorta results in a dramatic difference in flow kinetics in the descending aorta, which may modify the flow characteristics of delivery of blood to the mesenteric circulation. Echocardiographic evaluation of the descending aorta demonstrates a lower aortic pulsatility in association with NEC, suggesting the possibility of an inherent abnormality of the systemic vasculature, which may be a factor in predisposing to NEC in the postoperative HLHS population (39).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


