20

Idiopathic Interstitial Pneumonias
Diffuse interstitial lung disease is a generic term encompassing a broad range of largely unrelated conditions that share the propensity to cause breathlessness or cough associated with bilateral abnormal opacities of various types on conventional chest radiographs or computed tomography (CT) scans. The idiopathic interstitial pneumonias are a subset of diffuse interstitial lung diseases characterized by expansion of the interstitial compartment (i.e., that portion of the lung parenchyma sandwiched between epithelial and endothelial basement membranes) by an infiltrate of inflammatory cells. The inflammatory infiltrate is sometimes accompanied by fibrosis, in the form of either abnormal collagen deposition or proliferation of fibroblasts capable of collagen synthesis.
Averill Liebow1,2 pioneered the notion that morphologic characteristics are useful in categorizing idiopathic interstitial pneumonias into clinically and histologically distinct groups. Other early investigators also demonstrated prognostically different subsets linked to differences in morphology.3–6 Liebow’s categories of usual interstitial pneumonia (UIP) and desquamative interstitial pneumonia (DIP) have persisted as important histologic groups, whereas bronchiolitis obliterans with classical interstitial pneumonia (BIP) and giant cell interstitial pneumonia (GIP) have disappeared from more recent classification schemes (Table 20–1).7–9 Idiopathic bronchiolitis obliterans with organizing pneumonia (BOOP), also referred to as cryptogenic organizing pneumonia (COP), differs in many clinical and morphologic aspects from other conditions in this category and is considered elsewhere (see Chapter 21). The relationship between lymphoid interstitial pneumonia (LIP) and the idiopathic interstitial pneumonias is controversial and it is reviewed with other primary pulmonary lymphoproliferative disorders (see Chapter 19).
The principles developed by Liebow are relevant regardless of the specific classification scheme to which one subscribes. He was careful to say that these were histologic patterns rather than free-standing diagnostic entities, and that each could occur in a variety of clinical contexts.1 For example, although UIP usually occurs in patients with idiopathic pulmonary fibrosis (IPF), it occasionally represents lung involvement attributable to an underlying systemic connective tissue disease, asbestosis, or drug-induced pulmonary disease (Table 20–2). Regardless of the clinical context, however, he maintained that histologic classification of interstitial pneumonias provides “clues both to etiology and to pathogenesis and certainly to natural history and prognosis.”1 In other words, although histologic patterns may not be free-standing diagnostic entities until linked to specific clinical circumstances, each significantly limits the differential diagnosis in terms of potential etiologies or associations and each has implications for treatment and prognosis. In the setting of a patient with unexplained (i.e., idiopathic) interstitial lung disease, these histologically defined patterns are indeed specific diseases. In this clinical context biopsy is usually intended not only to confirm the suspicion of an interstitial pneumonia but also to exclude various mimics such as sarcoidosis, hypersensitivity pneumonia (i.e., extrinsic allergic alveolitis), and pulmonary Langerhans’ cell histiocytosis. It is in this very setting that morphologic classification has proven to be a powerful tool in predicting prognosis.
| International Classification8 | |
|---|---|---|
Katzenstein and Myers7 | Clinical Diagnosis | Pathologic Pattern |
Usual interstitial pneumonia (UIP) | Idiopathic pulmonary fibrosis (IPF) | Usual interstitial pneumonia (UIP) |
Desquamative interstitial pneumonia (DIP) | Desquamative interstitial pneumonia (DIP) | Desquamative interstitial pneumonia |
Respiratory bronchiolitis interstitial lung disease (RBILD) | Respiratory bronchiolitis interstitial lung disease (RBILD) | Respiratory bronchiolitis |
Acute interstitial pneumonia (AIP) | Acute interstitial pneumonia (AIP) | Diffuse alveolar damage (DAD) |
Nonspecific interstitial pneumonia (NSIP) | Nonspecific interstitial pneumonia (NSIP) | Nonspecific interstitial pneumonia (NSIP) |
| Cryptogenic organizing pneumonia (COP) | Organizing pneumonia (OP) |
| Lymphoid interstitial pneumonia (LIP) | Lymphoid interstitial pneumonia (LIP) |
Usual Interstitial Pneumonia (UIP)/Idiopathic Pulmonary Fibrosis (IPF)
An American Thoracic Society (ATS) consensus statement defines IPF as “a specific form of chronic fibrosing interstitial pneumonia limited to the lung and associated with the histologic appearance of usual interstitial pneumonia (UIP) on surgical (thoracoscopic or open) lung biopsy.”10 In other words, the clinical term IPF should be restricted to those patients with idiopathic interstitial lung disease characterized by the pathologic lesion referred to as UIP. IPF/UIP is the most common of the idiopathic interstitial pneumonias, accounting for over 60% of cases in a retrospective review of 104 patients who underwent surgical lung biopsy.11 UIP, as mentioned previously, occasionally occurs in patients with other underlying conditions or etiologies (Table 20–2). In most patients, however, UIP and IPF are interchangeable terms.
Condition | Distinction from UIP/Idiopathic Pulmonary Fibrosis (IPF) |
|---|---|
Connective tissue diseases | History and supportive laboratory data |
Rheumatoid arthritis |
|
Systemic sclerosis |
|
Polymyositis/dermatomyositis |
|
Asbestosis | History |
| Asbestos bodies (iron stain) |
Drug-induced lung disease | History |
Chlorambucil |
|
Cyclophosphamide |
|
Lomustine |
|
Nitrofurantoin |
|
Pindolol |
|
Hermansky-Pudlak syndrome | History (oculocutaneous albinism, bruising/bleeding) |
| Pigmented (“ceroid”) alveolar histiocytes |
| Vacuolated (foamy) type 2 pneumocytes |
Dyskeratosis congenita | History (reticulate skin pigmentation, nail dystrophy, hypotrichosis, mucosal leukoplakia) |
Clinical Features
Patients with IPF usually present in the sixth or seventh decade of life with slowly progressive dyspnea and non-productive cough. Most cases are sporadic, although rare familial examples have been described.12–17 IPF does not occur in children. Men are affected more commonly than women are by a ratio of nearly 2:1.18 Physical findings include bibasilar inspiratory crackles, a nonspecific but characteristic finding in nearly all patients. Digital clubbing occurs in about half of patients. Laboratory abnormalities are mild and relatively nonspecific. Rheumatoid factor and antinuclear antibodies are present in a substantial minority of patients who lack other evidence of systemic connective tissue disease. Pulmonary function studies show a restrictive pattern (i.e., reduced lung volumes with relative preservation of airflow) accompanied by a reduction in the diffusion capacity for carbon monoxide (DLCO) with hypoxemia at rest and/or with exercise. Lung volumes are preserved in some smokers who have concomitant emphysema.19–21 Isolated reduction in DLCO with normal lung volumes also occurs in patients with early disease.22
Conventional chest radiographs show relatively non-specific findings that include diffuse interstitial opacities associated with a bibasilar distribution and reduced lung volumes (Fig. 20–1). High-resolution computed tomography (HRCT) scans have greatly improved the diagnostic accuracy of imaging techniques and show a characteristic pattern of peripheral (subpleural), irregular linear (“reticular”) opacities involving upper and lower lung zones associated with architectural distortion in the form of traction bronchiectasis and bibasilar honeycomb change (Fig. 20–2).23–37 This relatively specific combination of radiologic findings is present about half the time, allowing accurate diagnosis by experienced observers in 40 to 50% of cases.38–41 The converse is that at least half of patients lack characteristic changes on imaging studies, and it is in this group of patients that surgical lung biopsy is most useful.
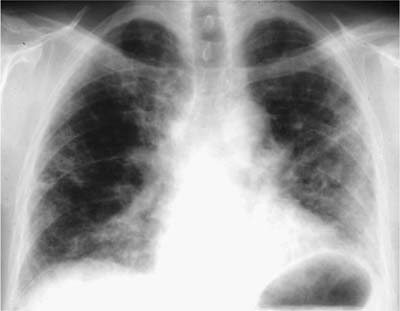
FIGURE 20–1 Conventional posteroanterior (PA) chest radiograph showing changes typical of usual interstitial pneumonia (UIP). Lung volumes are reduced, and there are linear opacities in the lower lung zones.
IPF usually follows a relentlessly progressive course with most patients dying of respiratory failure within 2 to 5 years of diagnosis.11,42–50 Severe abnormalities in measures of pulmonary function [i.e., forced vital capacity (FVC), DLCO, oxygen desaturation with exercise] at time of diagnosis are associated with a worse prognosis.49–56 Symptom progression, loss of pulmonary function, and progression of radiologic infiltrates within the first year of diagnosis are also associated with shorter survival.57–59 Periodic exacerbations associated with accelerated decline in symptoms and pulmonary function are the rule and can be related to either variation in the tempo of the underlying lesion or superimposed complications of various types. The term accelerated or acute exacerbation of IPF has been applied to patients with sudden worsening of symptoms over a period of a few weeks associated with new diffuse infiltrates on chest radiographs, sudden decrease in PaO2 resulting in acute respiratory failure, and absence of an identifiable infection or underlying heart failure.60–63 Most affected patients do not survive acute exacerbation of IPF/UIP.64,65 Respiratory failure is the most common cause of death overall in patients with IPF.66 Other complications that contribute to IPF-associated mortality include heart failure, bronchogenic carcinoma, ischemic heart disease, infection, and pulmonary embolism.66 Lung carcinoma is an important and frequently fatal complication of UIP affecting principally older men with a history of cigarette smoking.67–73 Peripheral squamous cell carcinomas comprise a disproportionate percentage of tumors in this population.67
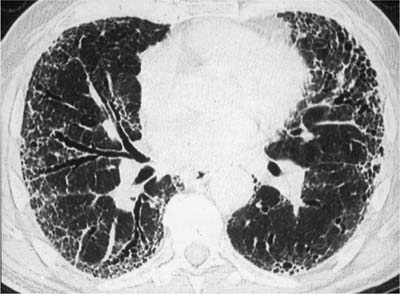
FIGURE 20–2 High-resolution computed tomography (CT) scan demonstrating features typical of UIP. Reticulonodular infiltrates are present in peripheral lung zones affecting mainly the posterior portions of the lower lobes. Traction bronchiectasis and honeycomb change are important indicators of lung fibrosis.
Corticosteroids are the historical standard therapy for IPF and are often administered in combination with another immunosuppressive agent, most commonly azathioprine or cyclophosphamide.10,74–76 The previously referenced ATS consensus statement acknowledges immunosuppressive agents as the standard of care despite the absence of any conclusive evidence of efficacy.10 Evolution in our understanding of pathogenesis has generated interest in novel treatment strategies (e.g., pirfenidone, interferon-γ) targeting various cytokines participating in the fibrogenic cascade, but to date none has emerged as standard therapy.74,77–80 Lung transplantation is used in some patients, but older age and comorbidities often exclude patients from consideration.81–85
Pathologic Features
Usual interstitial pneumonia is a specific morphologic entity. Whole lungs demonstrate honeycomb change affecting predominantly peripheral lower lung zones (Fig. 20–3). The histologic hallmark and chief diagnostic criterion in surgical lung biopsies is a heterogeneous or variegated appearance with alternating areas of normal lung, interstitial inflammation, fibrosis, and honeycomb change (Fig. 20–4). Alternating areas of abnormality that differ in character result in a distinctive “patchwork” appearance at low magnification (Fig. 20–5).86 Peripheral subpleural parenchyma is most severely affected, although a peripheral subpleural distribution may not be appreciable in some surgical lung biopsies.
Fibrosis predominates over inflammation in classical UIP.7,8,42–45,47,87 Fibrotic zones are composed mainly of dense eosinophilic collagen. Associated foci of fibroblast proliferation (“fibroblast foci”) are a consistent and important finding (Fig. 20–6).7,8,45 Fibroblast foci are randomly distributed within areas of interstitial collagen deposition and comprise fibroblasts and myofibroblasts arranged in a linear fashion within a pale staining matrix. Overlying epithelium consists of hyperplastic pneumocytes or columnar nonciliated bronchiolar cells. Fibroblast foci represent microscopic zones of acute lung injury (see Pathogenesis, later in this chapter) set against a backdrop of chronic scarring, thus contributing to the variegated appearance or temporal heterogeneity typical of UIP. Fibroblast foci are not specific for UIP, but are invariably present and therefore represent an important diagnostic criterion. Osseous metaplasia is often seen in areas of fibrosis and in uncommon cases can be extensive, likely accounting for at least some descriptions of diffuse or “dendriform” ossification. Honeycomb change is present in most surgical lung biopsies and is an important diagnostic feature. Honeycomb change is defined by cystically dilated bronchioles lined by columnar respiratory epithelium in scarred, fibrotic lung tissue (Fig. 20–7). Cystic honeycomb spaces often contain a combination of mucus and inflammatory cells. Fibrotic scars resulting in obliteration of the underlying lung architecture without associated honeycomb change are another form of architectural distortion characteristic of UIP. Smooth muscle hyperplasia is commonly seen in areas of fibrosis and honeycomb change and can be striking in some cases.
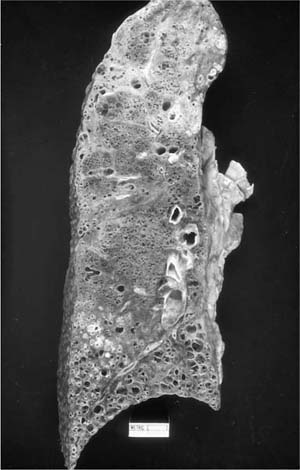
FIGURE 20–3 Whole autopsy lung showing UIP with associated bronchopneumonia. There is honeycomb change extensively involving the lung bases and peripheral, subpleural parenchyma with relative sparing of upper and central lung zones.
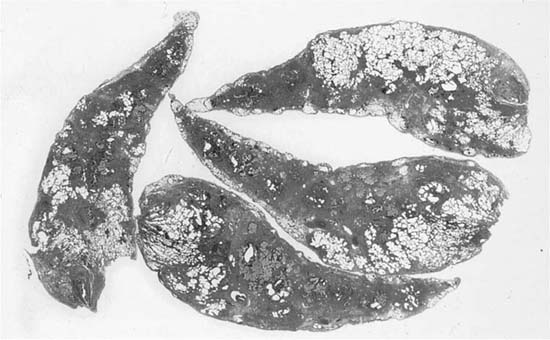
FIGURE 20–4 Whole mount of surgical lung biopsy showing the patchy, heterogeneous distribution of abnormalities typical of classical UIP.
Interstitial inflammation is relatively mild in most cases of UIP, consisting of a patchy alveolar septal infiltrate of lymphocytes, plasma cells, and histiocytes (Fig. 20–8). Associated peribronchiolar lymphoid aggregates occur in some patients but are rarely a dominant feature. Airspaces may contain clusters of lightly pigmented (“smokers’”) histiocytes resulting in foci that superficially resemble DIP (“DIP-like” foci) (Fig. 20–9).86 DIP-like changes are common and serve only as markers of cigarette smoking rather than evidence of a meaningful link between UIP and DIP. Another sometimes confusing finding is the presence of patchy areas in which airspaces contain a combination of histiocytes and eosinophils, thus resembling eosinophilic pneumonia (Fig. 20–10). Against a backdrop of otherwise typical UIP, “eosinophilic pneumonia-like” areas have no etiologic or clinical significance.88
Various forms of epithelial hyperplasia accompany the inflammatory and fibrotic changes in UIP.89 Thickened alveolar septa are lined by hyperplastic type 2 pneumocytes (Fig. 20–8). Hyperplastic pneumocytes occasionally contain cytoplasmic filaments analogous to Mallory’s hyaline more typically associated with hepatocytes in various forms of hepatitis (Fig. 20–11). This cytopathic change is not specific, however, and can be seen in other conditions such as diffuse alveolar damage (DAD) and asbestosis.90,91 Peribronchiolar interstitium participates in the inflammatory and fibrotic process, resulting in a bronchiolocentric distribution in some fields. Affected airways often show hyperplasia of bronchiolar epithelium.92 The presence of airway-centered injury may be confusing and must be interpreted in the context of concomitant changes in more distal, peripheral parenchyma.
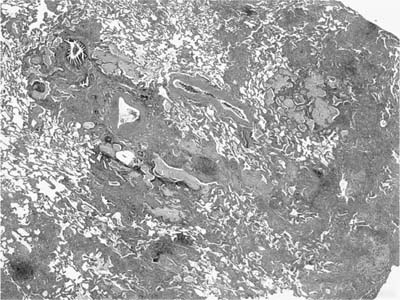
FIGURE 20–5 Low-magnification (original magnification ×20) photomicrograph showing patchwork distribution of abnormalities in UIP. In a single low-magnification field there are areas of uninvolved lung juxtaposed with areas of relatively mild interstitial thickening due to a combination of fibrosis and inflammation, and foci of honeycomb change. The honeycomb change consists of areas of early cystic change with associated mucostasis.
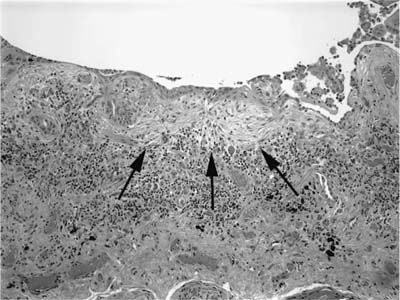
FIGURE 20–6 Photomicrograph (original magnification ×100) showing a fibroblast focus (arrows) in UIP. Against a backdrop of collagen fibrosis there is a subepithelial area of pallor in which spindle cells are arranged in a somewhat linear fashion in a pale-staining stroma. There is hyperplasia of the overlying epithelium.
Patients who are biopsied during a period of accelerated decline or acute exacerbation (“accelerated IPF”) often show a combination of UIP and superimposed DAD or organizing pneumonia.60,61 Indeed, DAD is frequently identified in autopsies of hospitalized patients with UIP, suggesting that this combination of findings may be a common terminal event.93 Evidence of superimposed DAD consists of areas in which alveolar septa are expanded by more extensive fibroblast proliferation than that seen in conventional fibroblast foci (Fig. 20–12). Areas of alveolar septal expansion are often associated with other features of acute lung injury, including marked hyperplasia and cytologic atypia in type 2 pneumocytes, hyaline membrane remnants, fibrin thrombi in small vessels, and squamous metaplasia of bronchiolar epithelium. In other patients the superimposed pattern of acute lung injury more closely resembles organizing pneumonia and is characterized by intraluminal plugs or polyps of fibroblastic tissue (Fig. 20–13). In any given patient organizing DAD and organizing pneumonia may show considerable histologic overlap. The presence of hyaline membranes is the single most useful feature in distinguishing the two.
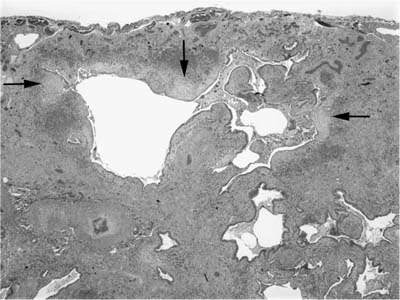
FIGURE 20–7 Low-magnification (original magnification ×20) photomicrograph showing honeycomb change in UIP. Cystically dilated spaces are present in an area of fibrosis adjacent to the visceral pleura. The cystic spaces are lined by columnar bronchiolar type epithelium. Multiple fibroblast foci are also present (arrows).
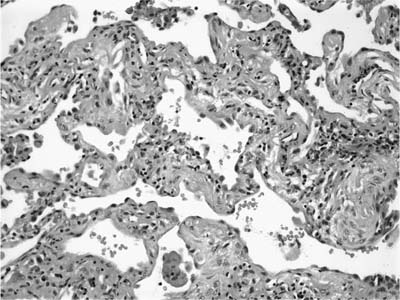
FIGURE 20–8 Photomicrograph (original magnification ×200) showing an area of interstitial pneumonia in UIP. Thickened alveolar septa contain a relatively mild infiltrate of mononuclear inflammatory cells and are lined by plump, hyperplastic pneumocytes.
No single histologic finding consistently predicts treatment response or prognosis in individual patients with UIP. Semiquantitative scoring systems have been used in research studies to correlate various histologic features with physiologic and radiologic measures of disease severity, but these have limited value in evaluating individual patients.20,94–97 Patients with more extensive fibroblast foci have experienced shorter mean survivals in some studies, whereas other investigators have failed to demonstrate the same relationship to outcome.98–100 Patients biopsied during an accelerated phase of illness in which organizing pneumonia or DAD is superimposed on underlying UIP may account, at least in part, for the link between extent of fibroblast proliferation and shortened survival observed in some studies.
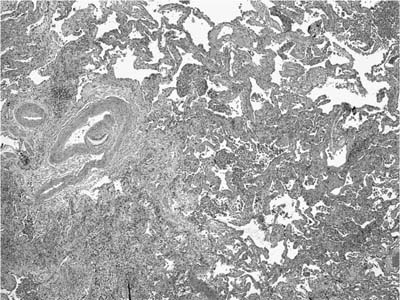
FIGURE 20–9 Low-magnification (original magnification ×40) photomicrograph showing desquamative interstitial pneumonia (DIP)–like changes in UIP. There is patchy fibrosis, most evident in the left portion of the field, associated with areas in which alveolar spaces contain clusters of pigmented histiocytes, resulting in an appearance resembling DIP.
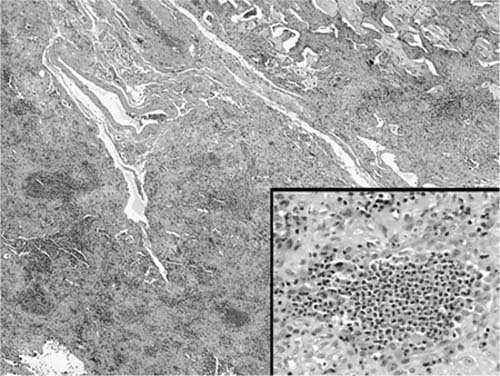
FIGURE 20–10 Low-magnification (original magnification ×40) photomicrograph showing an area of eosinophilic pneumonia in the lower left half of the field adjacent to an area of honeycomb change (upper right corner) in a patient with UIP. Higher magnification photomicrograph (inset; original magnification ×200) shows eosinophils filling an airspace.
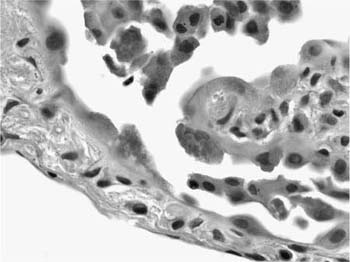
FIGURE 20–11 High-magnification (original magnification ×600) photomicrograph of UIP showing cytoplasmic hyaline in hyperplastic pneumocytes.
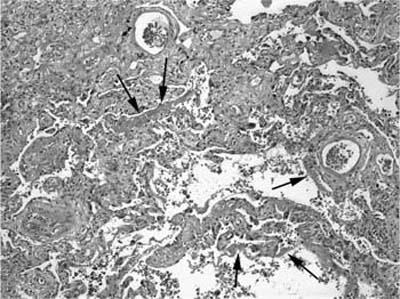
FIGURE 20–12 Photomicrograph (original magnification ×100) showing acute diffuse alveolar damage in a patient with UIP. Thickened alveolar septa are associated with well-developed hyaline membranes (arrows) in an 82-year-old man with an established diagnosis of idiopathic pulmonary fibrosis (IPF) who was admitted with rapidly worsening dyspnea and respiratory failure, requiring mechanical ventilation. Elsewhere the biopsy showed changes typical of UIP, including honeycomb change.
Pathogenesis
The cause of UIP in patients with IPF is unknown. Several associations have been identified including cigarette smoking, gastroesophageal reflux disease, occupational exposure to wood and various occupation-related dusts, and Epstein-Barr or hepatitis C virus infection.10,101–107 It may be that no single etiologic agent serves as a common inciting event but rather that affected patients have common defects in reparative pathways as outlined below.
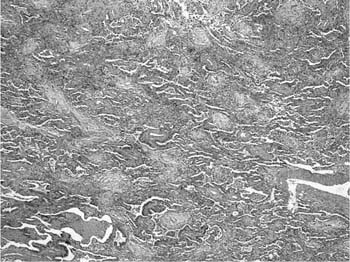
FIGURE 20–13 Photomicrograph (original magnification ×40) showing an area of organizing pneumonia in what was otherwise typical UIP in a patient with rapidly worsening shortness of breath (“accelerated IPF”).
A growing body of evidence indicates that epithelial injury and activation trigger a cascade of events resulting in fibrosis and honeycomb change in UIP.108–110 Fibroblast foci represent the sites of most recent epithelial necrosis and injury. Histopathologic, immunohistochemical, and ultrastructural studies have shown denudation and collapse of damaged epithelial basement membranes, migration of fibroblasts/myofibroblasts into airspaces, and extracellular matrix accumulation resulting in reorganized intraalveolar and interstitial compartments.108,111–116 These same events have been demonstrated at sites of acute lung injury in DAD and BOOP.111,113,115,117 The presence of these common events in very disparate clinical contexts suggests that defects in wound healing are responsible for differences in the pathogenesis of these conditions.
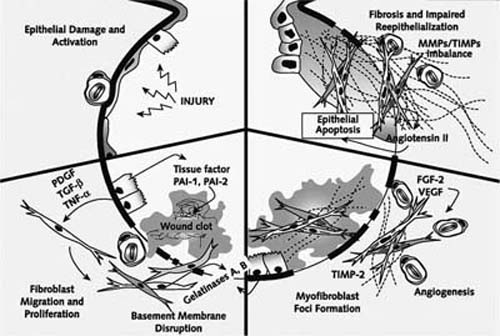
FIGURE 20–14 Summary of events in abnormal wound healing, extracellular matrix accumulation, and remodeling in UIP. Unknown injury initiates epithelial injury (upper left), which in turn induces antifibrinolytic activity in alveolar spaces. Damaged and reparative pneumocytes secrete antifibrinolytic factors while also generating various growth factors that induce migration, proliferation, and differentiation of fibroblasts and myofibroblasts (lower left). Epithelial cells and myofibroblasts elaborate gelatinases that may play a role in maintenance of basement membrane disruption, allowing migration of fibroblasts and myofibroblasts into the alveolar space and impeding reepithelialization (lower right). Intraalveolar and interstitial fibroblasts and myofibroblasts secrete matrix proteins, mainly collagens, which may be maintained and abnormally upregulated by an imbalance between collagenases and tissue inhibitors of metalloproteinases (upper right). Extra-cellular matrix accumulation is accompanied by angiogenesis related to elaboration of various angiogenic cytokines. FGF-2, fibroblast growth factor-2; MMP, metalloproteinase; PAI-1, PAI-2, plasminogen activator inhibitor-1, -2; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β; TIMP, tissue inhibitors of metalloproteinases; TNF-α, tumor necrosis factor-α; VEGF, vascular endothelial growth factor. (From Selman M, King TJ, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–151, with permission.)
Epithelial cells play an important role in early and late events that follow acute lung injury (Fig. 20–14).108,109,112,114,118–124 Following epithelial injury, surviving and regenerating respiratory epithelium contributes to abnormal antifibrinolytic activity at sites of injury resulting in persistence of an extracellular matrix that may impede wound healing. Tissue factor and plasminogen activator inhibitor-1 and -2 are elaborated by alveolar epithelial cells and contribute to this early antifibrinolytic activity. Alveolar pneumocytes also elaborate various fibrogenic cytokines and growth factors, including platelet-derived growth factor, transforming growth factor-β1, and tumor necrosis factor-α, important in recruitment and phenotypic modulation of fibroblasts and myofibroblasts.120,125,126 Nonepithelial inflammatory cells are also sources of proinflammatory and fibrogenic cytokines.127 Myofibroblasts in the sites of acute lung injury (i.e., fibroblast foci) amplify alveolar damage by inducing epithelial cell apoptosis mediated through angiotensinogen and associated with upregulation of p53 and p21Waf1/Cip1/Sdi1 in hyperplastic pneumocytes. Fibroblasts at acute lung injury sites may also fail to secrete hepatocyte growth factor, a cytokine with antifibrotic activity.128 Subepithelial myofibroblasts further contribute to basement membrane disruption in fibroblast foci through synthesis and secretion of gelatinases. Extracellular matrix accumulation and architectural remodeling results, at least in part, from an imbalance between collagenases and tissue inhibitors of metalloproteinases.116,129,130 Extracellular matrix accumulation is accompanied by angiogenesis; angiogenesis and vascular remodeling may in turn contribute to arteriovenous shunting.122,131 Despite the previously mentioned morphologic overlap, fibroblast foci of UIP differ from intraluminal fibroblast proliferation (Masson bodies) in BOOP in several important ways including fibroblast/myofibroblast phenotype, rates of fibroblast/myofibroblast apoptosis, modulation of extracellular matrix deposition, extent of neovascularization, character and extent of reepithelialization, and β-catenin expression. Upregulation of epithelial cell apoptosis and impaired adhesion to damaged basement membranes contributes to disordered reepithelialization at sites of acute lung injury in UIP. Differences between alveolar and bronchiolar epithelium in cell-cycle regulation and activation of the Wnt/β-catenin pathway may also contribute to architectural remodeling and honeycomb change.118,119
Differential Diagnosis
UIP is the most common of the idiopathic interstitial pneumonias and is unique in that the histologic findings are sufficiently distinctive to allow a specific clinical diagnosis to be made with a high level of accuracy in pathologically classical cases. Distinguishing UIP in patients with other underlying or associated conditions (Table 20–2) is largely a matter of correlation with clinical information. The presence of asbestos bodies is diagnostic of asbestosis in a patient with a well-documented history of exposure (see Chapter 24). Cytoplasmic vacuolization in hyperplastic pneumocytes coupled with pigmented alveolar histiocytes is a helpful histopathologic clue to the diagnosis of Hermansky-Pudlak syndrome, a condition restricted to patients with oculocutaneous albinism.132–140 Pathologic findings helpful in distinguishing UIP from other idiopathic interstitial pneumonias are detailed in discussions of individual entities. Areas indistinguishable from DIP and nonspecific interstitial pneumonia (NSIP) sometimes occur focally in otherwise typical UIP.86 For that reason correlation with radiologic findings is important in diagnostically challenging cases to minimize the impact of sampling bias.
Hypersensitivity pneumonia (also termed extrinsic allergic alveolitis), Langerhans’ cell histiocytosis (LCH), and BOOP are other potential mimics of UIP. Details concerning each of these entities are available elsewhere (see Chapter 21). Briefly, hypersensitivity pneumonia tends to be a more cellular and bronchiolocentric process. Peribronchiolar infiltrates in hypersensitivity pneumonia demonstrate varying degrees of granulomatous inflammation in most cases, usually in the form of isolated multinucleated giant cells or ill-defined clusters of epithelioid histiocytes. Honeycomb change can occur in hypersensitivity pneumonia but typically is not associated with the same degree of peripheral subpleural fibrosis.
Langerhans’ cell histiocytosis also tends to have a distinctly bronchiolocentric distribution. The bronchiolocentric nodules of LCH are frequently stellate in configuration and show variable degrees of fibrosis. Cases in which fibrotic nodules predominate may be difficult to distinguish from UIP. The key features are the stellate configuration and bronchiolocentric distribution of the nodules in LCH coupled with often striking paracicatricial airspace enlargement (“scar emphysema”). Fibroblast foci are also uncommon in purely fibrotic nodules in LCH. HRCT scans can be extremely useful in distinguishing LCH from UIP in diagnostically challenging cases.
Organizing pneumonia is a nonspecific pattern of lung injury that often occurs in combination with various unrelated pathologic processes including UIP. Idiopathic organizing pneumonia (i.e., BOOP) is a specific syndrome in which patchy intraluminal fibrosis occurs as a primary pathologic process. The absence of interstitial pneumonia or fibrosis away from zones of intraluminal fibrosis is an important feature in distinguishing BOOP from UIP.
Desquamative Interstitial Pneumonia (DIP)/Respiratory Bronchiolitis Interstitial Lung Disease (RBILD)
DIP and RBILD are two highly related, and in some cases indistinguishable, forms of diffuse interstitial lung disease typically grouped with the idiopathic interstitial pneumonias. Despite historical suggestions that DIP was merely an early form or precursor of UIP, current authorities agree that they represent unrelated forms of interstitial pneumonia with substantial clinical and histopathologic differences.7,8,45,47 DIP is relatively uncommon, accounting for 8 to 17% of biopsied patients with idiopathic interstitial pneumonias.11,45,47,141
Liebow et al2 noted a patchy and distinctly bronchiolocentric distribution of the histologic abnormalities in patients with mild or early DIP, and illustrated the lesion for which Niewoehner and colleagues142 subsequently coined the term respiratory bronchiolitis. Niewoehner et al described respiratory bronchiolitis as a form of small airway disease in autopsies of cigarette smokers who were the victims of sudden nonhospital deaths, and were primarily interested in the extent to which respiratory bronchiolitis might contribute to airflow limitation in smokers.142 Since then respiratory bronchiolitis and changes resembling those seen in DIP (“DIP-like” changes) have been recognized as markers of cigarette smoking that frequently occur as incidental findings in surgical and autopsy lung specimens.143–145 As originally noted by Liebow et al, however, a subset of patients with respiratory bronchiolitis have a syndrome of diffuse interstitial lung disease (i.e., RBILD) with many of the clinical and histologic attributes of DIP.146–153 RBILD is an uncommon form of interstitial pneumonia, accounting for only 2% of biopsied Mayo Clinic patients originally suspected of having IPF.11
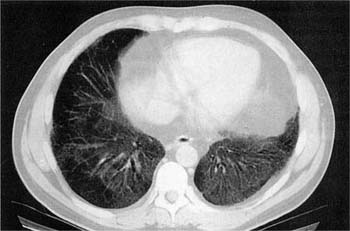
FIGURE 20–15 High-resolution CT scan in desquamative interstitial pneumonia. There is a confluent (left) and patchy (right) basilar ground-glass change in both lungs in this biopsy-confirmed case of desquamative interstitial pneumonia.
Clinical Features
DIP typically affects younger patients, with a mean age at diagnosis of 44 years compared with 59 years for UIP.2,42,45,47,141,153–155 DIP also differs from UIP in that it occasionally presents in childhood.156–166 Cigarette smokers comprise the majority of adult patients, prompting many to consider DIP a form of smoking-related interstitial lung disease.148,149,151,152 Physiologic testing usually shows mild reduction in lung volumes associated with a moderate decrease in diffusing capacity.2,42,153–155,167 Radiologic abnormalities tend to be less severe than those seen in UIP.2,42,154,168 Conventional chest radiographs are normal in as many as one fifth of patients.42,153 HRCT shows patchy ground-glass opacities, often with a lower lung zone distribution, without the honeycomb change characteristic of UIP (Fig. 20–15).24,153,169–171 In two separate studies of patients who had serial CT scans over the course of their illness, no “progression” from DIP to UIP was documented, further supporting the notion that DIP and UIP are separate and distinct entities.172,173
RBILD occurs almost exclusively in heavy cigarette smokers, usually in the fourth or fifth decade of life with average exposures of 35 pack-years of cigarette smoking.146,150,153 RBILD has not been reported in children. Men are affected more often than women. Rare examples have been reported in never-smokers with other occupationally related fume exposures.146 Symptoms are usually mild and include the insidious onset of dyspnea and a new or changed cough. Conventional chest radiographs are abnormal in about 80% of patients and typically show diffuse fine reticular or reticulonodular opacities in a bibasilar distribution (Fig. 20–16A).150,153 Patchy ground glass attenuation is the most frequently observed abnormality on CT scans, overlapping with the nonspecific combination of radiologic findings in DIP (Fig. 20–16B).169,174,175
DIP and RBILD are both associated with a significantly better prognosis than UIP. Overall survival in published studies of DIP is nearly 90%, ranging from around 70 to 80% in older studies to 100% in recently published series. To some extent the higher survival rates in recently published studies reflects a tendency to arbitrarily assign cases with associated fibrosis to the category of NSIP. Occasional patients spontaneously recover. Smoking cessation is an important therapeutic intervention in those who smoke. Corticosteroids with or without other immunosuppressive agents are beneficial in the majority of treated patients. Late relapses have been described after prolonged periods of recovery in a few patients.176,177 Recurrence in lung allografts has been reported after transplantation.178,179 RBILD is a relatively benign self-limited condition. Most patients either improve after smoking cessation or have stable, nonprogressive disease. None of the reported patients have died of their lung disease.

FIGURES 20–16 Respiratory bronchiolitis-associated interstitial lung disease. (A) Chest radiograph shows fine bilateral interstitial infiltrates. (B) High-resolution CT scan shows bilateral patchy increased attenuation (ground-glass change).
Pathologic Features
DIP differs histologically from UIP in that the changes tend to be much more uniform at low magnification without the variegated appearance typical of UIP (Fig. 20–17).1,2,7,8 The most striking feature is the presence of numerous pigmented alveolar macrophages, including occasional multinucleated cells, within distal airspaces. Liebow originally speculated that the pigmented cells might instead be desquamated pneumocytes, but multiple investigators have demonstrated that nearly all are macrophages distributed as relatively cohesive intraalveolar clusters.167,180–183 Small numbers of eosinophils and lymphocytes are also present. The macrophages are distinctive in that they have abundant cytoplasm, usually containing finely granular dusty brown pigment (Fig. 20–18). The cytoplasmic granules represent complex phagolysosomes that stain faintly with Prussian blue iron stains. Periodic acid-Schiff (PAS) stains also highlight the cytoplasmic granules after diastase predigestion, but this is a very nonspecific phenomenon of limited diagnostic value.
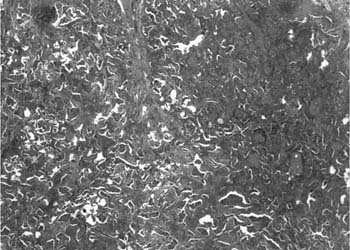
FIGURE 20–17 Low-magnification photomicrograph (original magnification ×20) showing DIP. There is uniform and diffuse alveolar septal thickening without honeycomb change or scarring. Alveolar spaces are filled with clusters of histiocytes, resulting in a consolidated or solid appearance.
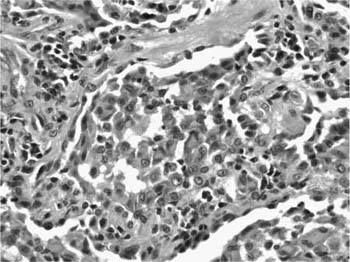
FIGURE 20–18 High-magnification photomicrograph (original magnification ×400) showing lightly pigmented macrophages typical of DIP. Alveolar histiocytes contain abundant cytoplasm that is finely granular, a feature that can be highlighted with iron stains.
Recognition of DIP as a discrete histologic entity requires the presence of not only lightly pigmented alveolar histiocytes but also a concomitant interstitial pneumonia. Alveolar septa are thickened by a sparse inflammatory infiltrate that often includes plasma cells and occasional eosinophils (Fig. 20–19). Lymphoid hyperplasia is seen in the majority of cases and consists of peribronchiolar lymphoid aggregates with secondary germinal centers (Fig. 20–20).2 A uniform population of plump cuboidal pneumocytes lines thickened alveolar septa. Mild fibrosis in the form of collagen deposition is common, but extensive fibrosis is unusual. Occasional foci of fibroblast proliferation resembling the fibroblast foci more typical of UIP can occur, but they are not a prominent or consistent feature. Fibrosis in DIP is usually mild and uniformly expands alveolar septa and peribronchiolar interstitium without architectural distortion in the form of either honeycomb change or destructive scarring (Fig. 20–21). The absence of associated architectural distortion is a key feature in separating DIP from UIP.1,2,7,8,86
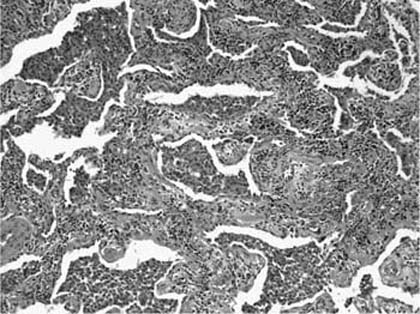
FIGURE 20–19 Photomicrograph (original magnification ×100) showing diffuse alveolar septal thickening in DIP. Alveolar septa are expanded by an infiltrate of mononuclear inflammatory cells with minimal associated collagen deposition. Airspaces are filled with histiocytes arranged in compact clusters.
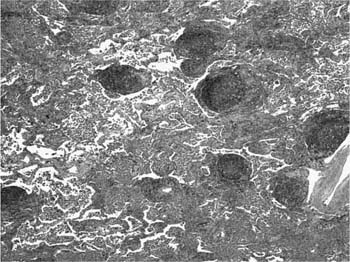
FIGURE 20–20 Low-magnification photomicrograph (original magnification ×40) illustrating lymphoid hyperplasia in DIP. Prominent lymphoid aggregates are present within bronchovascular bundles. To some extent the findings resemble those seen in follicular bronchiolitis (see Chapter 19), except that there is an associated interstitial pneumonia with features typical of DIP.
Respiratory bronchiolitis shares with DIP the presence of lightly pigmented macrophages as the most striking histologic feature.7,8,143,146,150,153 The changes are patchy at low magnification and have a distinctly bronchiolocentric distribution without the associated interstitial pneumonia necessary for a diagnosis of DIP (Fig. 20–22). Respiratory bronchioles, alveolar ducts, and peribronchiolar alveolar spaces contain clusters of the same dusty brown macrophages seen in DIP. The pigmented macrophages tend to lack the cohesive clustering typical of DIP. A mild submucosal and peribronchiolar infiltrate of lymphocytes and histiocytes expands the peribronchiolar interstitium. Interstitial histiocytes may contain cytoplasmic pigment identical to that seen within the intraluminal macrophages; coarse black anthracotic pigment is present in some patients. Mild peribronchiolar fibrosis is also seen and expands contiguous alveolar septa that are lined by hyperplastic type 2 cells and cuboidal bronchiolar-type epithelium. The combination of peribronchiolar interstitial thickening, epithelial hyperplasia, and pigmented intraluminal macrophages mimics the appearance of DIP (Fig. 20–23), and in any individual case the distinction may be arbitrary.
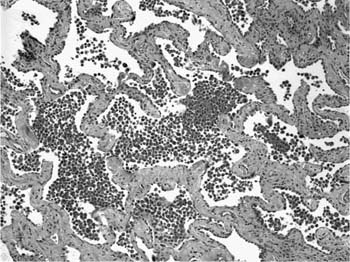
FIGURE 20–21 Photomicrograph (original magnification ×100) showing uniform alveolar septal fibrosis in DIP. Alveolar septa are expanded by deposition of eosinophilic collagen with minimal associated inflammation. Despite the fibrosis, the lung architecture is maintained without the honeycomb change or scarring typical of UIP.
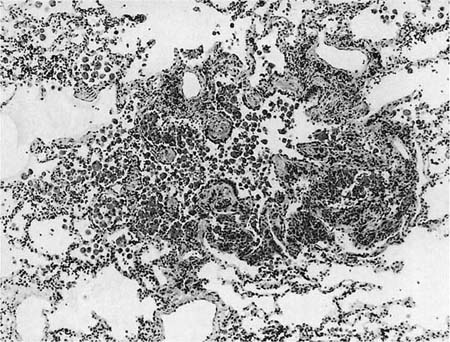
FIGURE 20–22 Respiratory bronchiolitis. The changes in this respiratory bronchiole were an incidental finding in unused donor lung tissue from a lung transplant. A respiratory bronchiole shows an increase in luminal macrophages and mural inflammation, with some extension of the inflammation and macrophages into the surrounding alveoli. There is slight interstitial fibrosis.
Pathogenesis
Both DIP and RBILD are associated with cigarette smoking; the association is stronger and more consistent in RBILD. DIP has also been described in patients with various occupational exposures (e.g., aluminum and asbestos), and in patients with drug-induced lung disease related to use of nitrofurantoin, sulfasalazine, and tocainide.180,184–186 Little is known about the specific events leading to fibrosis in DIP. Ultrastructural studies of lung histiocytes from smokers demonstrate increased numbers of cytoplasmic lysosomes and phagolysosomes. In some patients these cytoplasmic organelles contain plate-like crystalline inclusions composed of various metals (e.g., kaolin) contained in cigarette smoke.187,188 Marked increases in cytoplasmic iron and iron-binding proteins are also characteristic of smokers’ histiocytes.189 It is unknown how or if these alterations in alveolar macrophages spark the series of events that result in peribronchiolar and alveolar septal inflammation and fibrosis. The absence or paucity of fibroblast foci coupled with relative preservation of lung architecture suggests a pathogenesis that is different from that proposed for UIP. Comparisons with UIP have demonstrated differences in tenascin accumulation, cell cycle regulatory proteins, and Wnt/β-catenin pathway activation.118,119,121,123
Differential Diagnosis
Neither DIP nor RBILD should be viewed as free-standing histopathologic entities, because areas resembling both commonly occur in cigarette smokers with other lung diseases, including UIP. In a review of consecutive surgical specimens, Fraig and associates143 identified 109 surgical lung specimens with respiratory bronchiolitis that included “DIP-like” areas in six. Of these, only two patients had clinical syndromes of either DIP or RBILD. There are no histologic features that reliably distinguish patients with incidental respiratory bronchiolitis and DIP-like reactions from patients with the clinical syndromes of RBILD or DIP, respectively.143,147,190 Because the significance of respiratory bronchiolitis and DIP-like changes can be established only on the basis, of both the clinical and histologic findings, neither RBILD nor DIP can be diagnosed on small biopsy specimens. Indeed, given the dangers of sampling bias, DIP and RBILD should be diagnosed only when other forms of interstitial lung disease have been vigorously excluded, a process that requires correlation with clinical and radiologic findings.
Distinguishing UIP with “DIP-like” changes from DIP can be extremely difficult. Histologic distinction hinges on recognition of the characteristically variegated pattern of lung involvement with associated honeycomb change in UIP. To some extent the difficulty in making this distinction is linked to sampling bias, and for that reason correlation with radiologic findings can be extremely useful in difficult cases. Prominent DIP-like changes in a biopsy from a patient with radiologic findings typical of UIP on HRCT scan, for example, should not be interpreted as DIP.
Langerhans’ cell histiocytosis, a lesion seen almost exclusively in cigarette smokers, is often associated with respiratory bronchiolitis and “DIP-like” changes.191 The presence of distinctive bronchiolocentric nodules containing variable number of histiocyte-like Langerhans’ cells is key to the differential diagnosis (see Chapter 21). Eosinophilic pneumonia resembles DIP in that airspaces contain a cellular infiltrate of not only eosinophils but also variable numbers of alveolar macrophages, including occasional multinucleated giant cells (see Chapter 21). The number of eosinophils is greater in eosinophilic pneumonia and is often accompanied by a fibrinous exudate and eosinophilic abscesses. The airspace filling process in eosinophilic pneumonia tends to be more patchy in distribution. Absence of respiratory bronchiolitis away from areas of airspace consolidation is also helpful in distinguishing eosinophilic pneumonia from DIP.
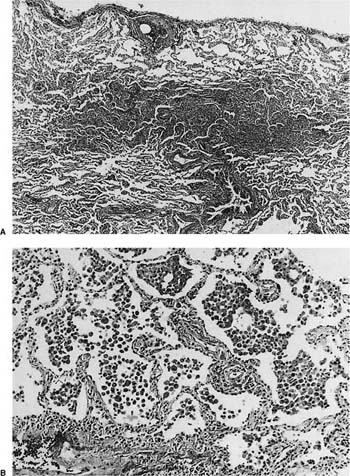
FIGURE 20–23 Respiratory bronchiolitis-associated interstitial lung disease. (A) Low power shows peribronchiolar consolidation with sparing of the surrounding alveoli. (B) Higher power shows fields indistinguishable from desquamative interstitial pneumonia, with increase in alveolar macrophages and mild inflammation and thickening of alveolar walls.
The presence of iron-containing cytoplasmic pigment in respiratory bronchiolitis and DIP may cause confusion with alveolar hemorrhage syndromes. The cytoplasmic pigment in DIP and RBILD tends to be more finely granular without the coarse refractile hemosiderin particles resulting from hemorrhage. Iron stains also tend to be more faintly positive in the cytoplasmic granules attributable to cigarette smoking, but this may vary according to the number of pack-years of exposure. Extracellular hemosiderin deposition, including encrustation of vascular elastica (“endogenous pneumoconiosis”), is common in hemosiderosis, resulting from recurrent alveolar hemorrhage and chronic venous hypertension, and does not occur in either DIP or RBILD.
Acute Interstitial Pneumonia (AIP)
Most idiopathic interstitial pneumonias are chronic processes characterized by an insidious or subacute onset and a slowly progressive course. A subset of patients present with acute, and sometimes explosive, onset of respiratory symptoms followed by a rapidly progressive course culminating in respiratory failure. Most patients in this category have so-called acute interstitial pneumonia (AIP), also referred to as Hamman-Rich disease.7,8,192 This acute variant of idiopathic interstitial pneumonia is analogous to acute respiratory distress syndrome (ARDS), differing from classical cases only in that it is not preceded by an identifiable catastrophic event.
Clinical Features
Patients with AIP present with rapidly evolving shortness of breath and nonproductive cough of 1 to 3 weeks’ duration, preceded by symptoms attributable to an upper respiratory tract infection.63 Men and women are affected equally with a mean age at diagnosis of 55 years, similar to that described for UIP but with a much broader age range that includes children.62,192–201 Fever is present in about half of patients. Most patients are severely hypoxemic at the time of diagnosis and require hospitalization and mechanical ventilation. Microbiologic assays and serologic studies, by definition, fail to identify an etiologic agent. CT scans demonstrate a combination of ground-glass attenuation and consolidation in a bilateral and symmetrical distribution.62,193,195–197,199,202
AIP has the same poor prognosis as ARDS, with a collective mortality of over 50% in reported series.62,192–201 Published mortality rates are similar to those associated with UIP but differ in that mean and median survival intervals are much shorter, ranging from 2 to 10 weeks. Higher survival rates have been observed in more recently published series.200 Some patients fully recover; others recover only to suffer multiple relapses.201 Chronic persistent interstitial lung disease occurs in a minority of survivors and may mimic NSIP (see below).201,203 Many patients are treated with corticosteroids, but there is no carefully controlled evidence that they are efficacious. Care is primarily supportive.
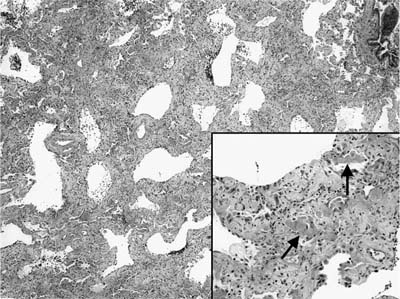
FIGURE 20–24 Low-magnification photomicrograph (original magnification ×40) showing the organizing phase of diffuse alveolar damage in a patient with acute interstitial pneumonia. Interstitium appears uniformly thickened without the variegated pattern typical of UIP. At higher magnification (inset; original magnification ×100) alveolar septa are collapsed on one another and thickened by fibroblast/myofibroblast proliferation. There is only a minimal associated infiltrate of mononuclear inflammatory cells. Remnants of hyaline membranes are also present (arrows).
Pathologic Features
Biopsies from patients with AIP demonstrate DAD, usually in the late or organizing stage, as described in greater detail elsewhere (see Chapter 14). There are no histologic features that reliably distinguish DAD in the setting of AIP from DAD of other known causes. For that reason a biopsy diagnosis of DAD should prompt a search for histologic clues to potential etiologies, especially those with treatment implications. Special stains, cultures, and serologic studies are especially helpful in vigorously excluding infectious causes.
Extensive fibroblast proliferation is the dominant finding in the organizing stage of DAD, and is usually conspicuous in biopsies from patients with AIP (Fig. 20–24).192,194,198 Alveolar septa are uniformly thickened and distorted by proliferating fibroblasts and myofibroblasts within a myxoid basophilic matrix. The uniformity of the findings contrasts sharply with the patchwork distribution of highly variegated abnormalities in UIP. Focally, the fibroblast proliferation may include intraluminal, polypoid structures indistinguishable from those seen in BOOP.198 Fibroblast proliferation is accompanied by marked hyperplasia of alveolar lining cells, often with associated cytologic atypia in the form of nuclear enlargement and prominent nucleoli. Cytoplasmic hyaline identical to the Mallory’s hyaline illustrated in UIP (Fig. 20–11) occurs in some patients and is of no special significance. Remnants of hyaline membranes are sometimes present but are rarely prominent (Fig. 20–24). Hyaline membranes, when present, are distributed along the surfaces of alveolar walls and ducts, and within remodeled alveolar septa. Multiple fibrin thrombi in small muscular arteries and squamous metaplasia of bronchiolar epithelium are other manifestations of acute lung injury that can be important clues to the diagnosis in difficult cases (Fig. 20–25). Extensive architectural remodeling may develop in as little as 3 to 4 weeks, resulting in honeycomb change characterized by cystically dilated bronchioles with concomitant collapse and fibrosis of distal lung parenchyma (Fig. 20–26). Honeycomb change complicating DAD is diffusely and uniformly distributed without the peripheral, subpleural, and bibasilar localization seen in UIP.
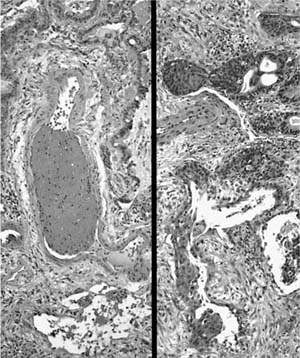
FIGURE 20–25 Photomicrographs (original magnification ×100) showing fibrin thrombus (left) and squamous metaplasia of bronchiolar epithelium (right) in surgical lung biopsies from patients with acute interstitial pneumonia.

FIGURE 20–26 Cut surface of inflated autopsy lung from patient who died about 2 months after the onset of acute interstitial pneumonia. There is extensive and uniformly distributed honeycomb change, without the bibasilar and peripheral distribution characteristic of UIP.
Pathogenesis
Ultrastructural studies of AIP have confirmed the presence of epithelial and endothelial damage. Epithelial necrosis is followed by denudation of subepithelial basement membranes, resulting in partial and complete collapse of alveolar septa.115 Alveolar collapse refers to wrinkling and invagination of denuded epithelial basal laminae. The invaginated segments become permanently apposed when the luminal surface is reepithelialized by proliferating type 2 pneumocytes. Total alveolar collapse occurs when the denuded basal laminae of adjacent alveolar septa become apposed and are likewise permanently fused by proliferating pneumocytes. In a process analogous to that described for UIP, fibroblasts and myofibroblasts migrate through gaps in damaged basement membranes into airspaces.113,115 Proliferation of reparative pneumocytes and bronchiolar epithelium results in mural incorporation of intraalveolar exudates. Mural incorporation occurs when intraalveolar exudates become sandwiched between proliferating type 2 cells and denuded alveolar septal basement membranes. Both alveolar collapse and mural incorporation of intraalveolar exudates contribute to the alveolar septal thickening seen by light microscopy.
Differential Diagnosis
Diffuse alveolar damage is the histologic counterpart of the ARDS and as such has a long list of potential causes (see Chapter 14). In nonimmunocompromised patients who lack a history of antecedent catastrophic injury, the main etiologic considerations are an as yet unidentified cause for DAD (e.g., influenza) and accelerated IPF (see earlier section on UIP in this chapter). Distinguishing the latter depends not only on recognizing underlying UIP but also on correlation with the clinical and radiologic findings. Histologic features useful in distinguishing DAD from organizing pneumonia (see Chapter 21) and NSIP (see later in this chapter) are detailed elsewhere.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


