Hypertrophic Cardiomyopathy
Rajeev Joshi
Mark Sherrid
Hypertrophic cardiomyopathy (HCM) is characterized by hypertrophy of the left ventricle that occurs without clinical causes such as hypertension or aortic valve disease and is marked by variable morphologic, clinical, and hemodynamic abnormalities. HCM is the great masquerader of cardiology. It often masquerades as coronary artery disease as it presents with anginal chest pain owing to myocardial ischemia—although in the absence of epicardial coronary narrowings. It is particularly confusing when HCM presents with ST depressions, T-wave inversions, or pathologic Q waves. It often masquerades as dilated or ischemic cardiomyopathy, with congestive heart failure and cardiomegaly on X-ray. It may masquerade as mitral or aortic valve disease with systolic murmur and heart failure symptoms. Sometimes, tragically, HCM can masquerade as normal, when young adults die suddenly, sometimes at rest and sometimes on the athletic playing field. With this great diversity of presenting syndromes, HCM patients are admitted to the cardiac care unit (CCU) not infrequently, where it is vital to rapidly diagnose them and initiate disease-specific therapy. Missing the diagnosis of HCM in the CCU can lead to disastrous outcomes.
Its pathophysiology, diagnosis, and treatment span the gamut of cardiologic disciplines. The pathophysiology includes understanding of left ventricular (LV) outflow obstruction, mitral regurgitation, ischemia, atrial fibrillation, sudden death, diastolic dysfunction, aspects of molecular biology and genetics. Diagnostic testing with echocardiography, nuclear scintigraphy, stress testing, catheterization, 24-hour ECG, and MRI may be applied. Treatment may involve pharmacologic agents, the implanted defibrillator, surgical septal myectomy, transcoronary intervention in the form of alcohol septal ablation (ASA), or pacing. However, when these lists are examined, one recognizes that this is identical to the spectrum encountered in more common cardiac diseases. The challenge in HCM is learning the disease-specific pathophysiology and treatment indications.
GENETICS
HCM is inherited as an autosomal dominant trait; approximately half of patients have another family member with HCM. Unexplained hypertrophy occurs in 1:500 in the general population, making it the most common inherited cardiac disorder. Sarcomeric mutations of 18 genes that code for myofilaments or their supporting proteins have been identified as a cause of HCM.1,2 In a cohort of referred, unrelated patients with HCM, approximately 50% of the patients were found to have sarcomeric mutations. In the remaining 50%, none of the known genotype abnormalities was found. Younger age at diagnosis, marked wall thickness, and a family history of HCM increase the frequency that a patient will be gene positive. Echocardiographic appearance also appears to predict a high likelihood of sarcomeric protein mutation HCM; a reversed septal curvature causing a crescent-shaped LV cavity predicts gene-positive patients as compared with those with localized subaortic bulge and preserved septal curvature.3 The most common mutations found are in the ß-myosin heavy chain and in myosin-binding protein C. Although most of genetically determined HCM occur on eight genes, many hundreds of HCM-causing mutations are dispersed over the many loci of the 18 genes. These genes may cause different phenotypes and may have different prognoses. Even among families with the same mutation on a particular locus, individuals vary with respect to phenotype and prognosis. This has markedly delayed genotype -phenotype correlation. The pathophysiologic linkage between mutations and hypertrophy appears to be mediated by mutation-induced functional abnormalities, most often because of increased contractile function. Recent theories about cause of hypertrophy have focused on inefficient utilization of ATP.
PATHOPHYSIOLOGY
On light microscopy individual myocyte hypertrophy is noted. Myocardial fiber disarray is the pathognomonic abnormality. In normal individuals, myocytes are arranged in linear parallel arrays. In HCM with fiber disarray, myocytes form chaotic intersecting bundles. With electron microscopy myofilament disarray is noted as well. Although fiber disarray is noted in other diseases, the percentage of the myocardium occupied by disarray is higher in patients with HCM. Fiber disarray is thought to predispose to electrical reentry and sudden death.
Fibrosis is also a prominent feature on light microscopy. Interstitial and perivascular fibrosis may occupy as much as 14% of the myocardium in patients who die suddenly. Fibrosis and hypertrophy decrease LV chamber compliance and cause diastolic dysfunction and exercise intolerance. Fibrosis appears to predispose to complex ventricular arrhythmia. Although the epicardial coronary arteries are dilated, narrowings of the intramural penetrating coronary arteries are noted, owing to arteriolar intimal and medial hyperplasia. These narrowings are thought to contribute to ischemia, well documented in HCM. Figure 12.1 shows such narrowings in myectomy resections.
DIAGNOSIS
HCM is diagnosed when LV hypertrophy occurs in the absence of a clinical condition that would cause the degree of hypertrophy noted.4,5 Thus, to make the diagnosis of HCM it is important to evaluate for other cardiac or systemic conditions capable of
producing the magnitude of hypertrophy evident (e.g., aortic valve stenosis, systemic hypertension, athlete’s heart. Athlete’s heart occurs only in elite highly trained competitive athletes where wall thickness up to 15 mm occurs.) Wall thickness >14 mm is the criteria used for diagnosis. Most of the patients who reach clinical attention have wall thicknesses between 20 and 30 mm. The location of the abnormal hypertrophy is most often the anterior septum, although the posterior septum and anterior wall are frequently hypertrophied as well. Typical of the heterogeneity of HCM is that hypertrophy can occur in any segment, even among relatives known to have the same genotype. Apical hypertrophy that spares the basilar and mid segments is a variant that occurs more frequently in East Asian patients with HCM. However, it is a relatively common variant in North American and European patients as well, occurring in 7%. This variant generally has a better prognosis. Truly atypical HCM variants include thickening just of the lateral wall or posterior wall. Wall thickening is most often assessed by two-dimensional echocardiography. Particular attention should be paid to the septum and also to the thickness of the anterior wall. The anterior wall is more difficult to visualize clearly than the septum because of poorer lateral resolution compared with the axial resolution of echocardiography systems. Magnetic resonance imaging may be useful in selected cases.
producing the magnitude of hypertrophy evident (e.g., aortic valve stenosis, systemic hypertension, athlete’s heart. Athlete’s heart occurs only in elite highly trained competitive athletes where wall thickness up to 15 mm occurs.) Wall thickness >14 mm is the criteria used for diagnosis. Most of the patients who reach clinical attention have wall thicknesses between 20 and 30 mm. The location of the abnormal hypertrophy is most often the anterior septum, although the posterior septum and anterior wall are frequently hypertrophied as well. Typical of the heterogeneity of HCM is that hypertrophy can occur in any segment, even among relatives known to have the same genotype. Apical hypertrophy that spares the basilar and mid segments is a variant that occurs more frequently in East Asian patients with HCM. However, it is a relatively common variant in North American and European patients as well, occurring in 7%. This variant generally has a better prognosis. Truly atypical HCM variants include thickening just of the lateral wall or posterior wall. Wall thickening is most often assessed by two-dimensional echocardiography. Particular attention should be paid to the septum and also to the thickness of the anterior wall. The anterior wall is more difficult to visualize clearly than the septum because of poorer lateral resolution compared with the axial resolution of echocardiography systems. Magnetic resonance imaging may be useful in selected cases.
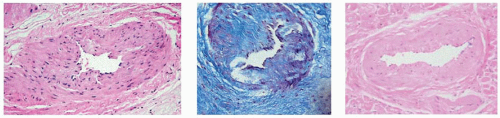 Figure 12.1. Histopathology from surgical specimens of three patients with obstructive HCM who underwent surgical septal myectomy for progressive heart failure symptoms. All three patients had intimal and medial hypertrophy of the intramural septal branches with luminal narrowing. Dense perivascular fibrosis is present in the middle frame. Left and right: Hematoxylin and eosin. Middle: Masson’s trichrome stain. |
In a subset of patients, the site and extent of cardiac hypertrophy and abnormalities of the mitral valve result in obstruction to LV outflow. LV outflow tract (LVOT) obstruction is an important determinant of symptoms and is associated with adverse outcome. The commonest location of obstruction is in the LV outflow tract, caused by systolic anterior motion (SAM) of the mitral valve and mitral-septal contact.6 The phenomenon of dynamic SAM is caused by a crucial anatomic overlap between the inflow and outflow portions of the left ventricle. Figure 12.2 shows dynamic SAM as it progresses though the early moments of systole. Narrowing of the LVOT and the anterior position of the coaptation point places the protruding leaflet into the edge of the flow stream, subject to the pushing force of flow that strikes the undersurface of the leaflet as illustrated in Figure 12.3.
LEFT VENTRICULAR OUTFLOW TRACT OBSTRUCTION
LVOT obstruction is quantified by measuring the pressure drop and the gradient across the narrowing. This is most commonly done with continuous wave Doppler echocardiography. Pulsed Doppler correlation with the two-dimensional echocardiogram allows determination of the site of obstruction, which must be ascertained in every patient, especially if intervention is contemplated. LV outflow obstruction causes a mid-systolic drop in LV ejection velocities and flow when the gradient is >60 mm Hg. This echocardiographic pattern has been termed the “lobster claw” abnormality because of its characteristic appearance (Figure 12.4). Resting obstruction is considered present when a resting gradient of 30 mm Hg is present. Reducing preload may provoke a gradient by increasing the overlap between the inflow and outflow portions of the LV. Patients who have no resting gradient but who have gradients >30 mm Hg after maneuvers have latent obstruction, and patients with mild obstruction <30 mm Hg that rises >30 mm Hg after maneuvers have provocable obstruction. Typically, Valsalva, exercise, and standing may be used to provoke obstruction7 and, on occasion, exercise in the postprandial state. As the main reason for provoking gradient is to correlate patient symptoms with obstruction and to provide a target for therapy, one should use only physiologic maneuvers, such as standing, exercise, or Valsalva. Dobutamine and amyl nitrite are not physiologic stimuli and should not be used to provoke gradient. Furthermore, dobutamine may provoke gradients in normal individuals. Clinically, we perform treadmill stress exercise echocardiography on all patients with HCM who are
able to exercise. An exception would be for patients with resting gradients of >80 mm Hg where little available information will be gained.
able to exercise. An exception would be for patients with resting gradients of >80 mm Hg where little available information will be gained.
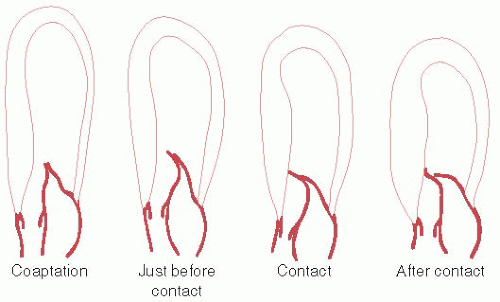 Figure 12.2. Systolic anterior motion of the mitral valve, drawn from apical five-chamber view, as it proceeds in early systole. (Adapted from Sherrid MV, Chu CK, Delia E, et al. An echocardiographic study of the fluid mechanics of obstruction in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1993;22:816-825.) |
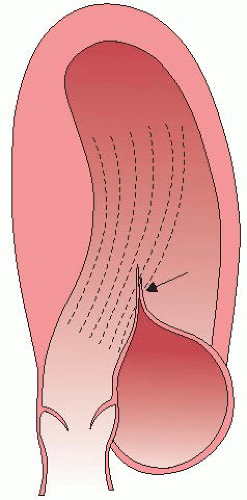 Figure 12.3. Left: The pushing force of flow. Intraventricular flow relative to the mitral valve in the apical five-chamber view. In obstructive HCM, the mitral leaflet coaptation point is closer to the septum than normal. The protruding leaflets extend into the edge of the flowstream and are swept by the pushing force of flow toward the septum. Flow pushes the underside of the leaflets (arrow). Note that the midseptal bulge redirects flow so that it comes from a relatively lateral and posterior direction; on the five-chamber view flow comes from “right field” or “1 o’clock” direction. This contributes to the high angle of attack relative to the protruding leaflets. Also note that the posterior leaflet is shielded and separated from outflow tract flow by the cowl of the anterior leaflet. Venturi flow in the outflow tract cannot be lifting the posterior leaflet because there is little or no area of this leaflet exposed to outflow tract flow. Venturi forces cannot be causing the anterior motion of the posterior leaflet. (Adapted from Sherrid MV, Gunsburg DZ, Moldenhauer S, et al. Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2000;36:1344-1354.) |
TREATMENT
There are five aspects of care that should be covered in the treatment of every HCM patient: (1) Risk of sudden death should be discussed and individual stratification of risk should be done. The average risk of dying suddenly is 1% per year for HCM patient populations, but the risk may be higher or lower depending on whether risk factors for sudden death are present. For patients deemed by their physicians to be at high risk, discussion of implantation of a cardioverter defibrillator should be considered. (2) Treatment of symptoms. (3) Discussion and advice to avoid athletic competition and extremes of exertion. (4) Detection and management of hypercholesterolemia (HCM and coronary disease have an adverse synergistic effect on prognosis). (5) Screening of first degree family members either through echocardiograms and EKGs, or through genetic testing to assess who has inherited the HCM genes. Although many aspects of this evaluation can wait until a patient is stabilized after the CCU stay, it must be remembered that HCM is a lifelong condition and that these issues need discussion and management when the dust clears.
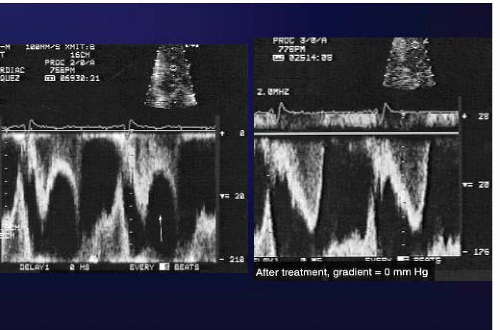 Figure 12.4. Left:
Get Clinical Tree app for offline access
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|