Hypertrophic Cardiomyopathy
Barry J. Maron
Cardiomyopathies, or primary diseases of the myocardium, are not uncommon in infants and children. Although these conditions have been the subject of intensive investigation, our understanding of this diverse group of diseases is constantly evolving with regard to clinical identification, genetics, natural history, and therapy. A contemporary diagnostic classification of cardiomyopathies, including those occurring in young patients, has been proposed through the American Heart Association (AHA) (1). One of the most important of these diseases to the practicing pediatric cardiology community is hypertrophic cardiomyopathy (HCM).
HCM is a genetic cardiac disease with heterogeneous clinical expression, unique pathophysiology, and a diverse natural history caused by mutations in 11 or more genes encoding proteins of the cardiac sarcomere (1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38).
HCM may be diagnosed and cause disability and death at virtually any age, including early childhood and infancy, and notably is the most common cause of sudden death (SD) in young people, including competitive athletes (4,5,39,40). On the other hand, HCM is also compatible with normal longevity often without disability or the need for major interventions to achieve that end (2,4,5,9).
Since its modern description >50 years ago (8), our understanding of the complexity of HCM has increased dramatically. On the other hand, perhaps no other childhood cardiovascular disease continues to present the challenges and controversies with respect to diagnosis, clinical course, and management as does HCM.
Definition and Nomenclature
HCM is characterized by a thickened but nondilated left ventricle (LV) in the absence of another cardiac or systemic disease capable of producing the magnitude of hypertrophy evident (e.g., aortic valve stenosis, systemic hypertension, or some physiologic expressions of athlete’s heart) (2,4,5,6,9).
In 1958, Donald Teare (8), the coroner of London, published the first modern pathologic report of this disease, in which he described SD in eight young people due to asymmetrical hypertrophy of the ventricular septum (VS) resembling a cardiac tumor. Subsequent investigations led to a dramatic evolution in our perceptions of the HCM clinical spectrum. In the process, the disease acquired a confusing array of names, all presumably describing the same clinical entity. Most of these terms used to describe HCM emphasized LV outflow obstruction. Thus, the names “idiopathic hypertrophic subaortic stenosis,” “hypertrophic obstructive cardiomyopathy,” and “muscular subaortic stenosis” (as well as their abbreviations: IHSS, HOCM, and MSS, respectively) became widely used. However, about one-third of patients with HCM have either no or only mild obstruction to LV outflow at rest or with exercise (2,3,4,5,23). Hence, the preferred name for this condition has become hypertrophic cardiomyopathy (with or without obstruction). However, it is potentially confusing to use this term to describe other systemic, metabolic, or multiorgan syndromes associated with increased LV wall thickness in infancy and childhood (21).
Prevalence
Several epidemiologic studies are now available showing that HCM is the most common of the genetic heart diseases occurring in at least 1:500 of the general population (2,24,25). This overall prevalence is disproportionate to the numerical recognition of HCM in clinical cardiology practice (26), particularly pediatric cardiology. HCM is a global disease now reported from >50 countries (27), with the most intense interest and reporting of cases from North America, Western Europe, Asia (Japan and China), and Australia.
Morphology
Left Ventricular Hypertrophy
Multiple echocardiographic studies have defined the gross morphologic features of HCM, particularly its pronounced diversity and myriad patterns of LV hypertrophy (LVH) (Figs. 52.1 to 52.3)
(2,3,4,5,6,7,16,17,18,41). Indeed, in HCM, there is no single or classic morphologic form, and virtually all possible patterns of LVH have been observed. Even closely related relatives with the same genetic substrate usually have dissimilar patterns of LVH, with the exception of identical twins in whom the hearts appear identical. Although truly symmetric (concentric) LVH may occur occasionally (6) (see Fig. 52.2), the distribution of wall thickening in HCM is characteristically asymmetric, in which ≥1 segment of the wall is thicker than other areas, including particularly heterogeneous patterns with contiguous segments of the LV wall differing greatly in thickness (7).
(2,3,4,5,6,7,16,17,18,41). Indeed, in HCM, there is no single or classic morphologic form, and virtually all possible patterns of LVH have been observed. Even closely related relatives with the same genetic substrate usually have dissimilar patterns of LVH, with the exception of identical twins in whom the hearts appear identical. Although truly symmetric (concentric) LVH may occur occasionally (6) (see Fig. 52.2), the distribution of wall thickening in HCM is characteristically asymmetric, in which ≥1 segment of the wall is thicker than other areas, including particularly heterogeneous patterns with contiguous segments of the LV wall differing greatly in thickness (7).
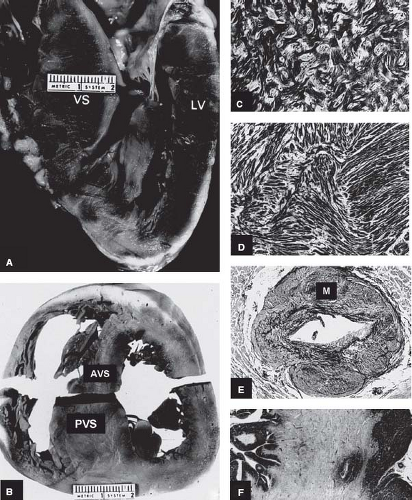 Figure 52.1 Morphologic components of the underlying disease process in HCM. A: A gross heart specimen sectioned in a cross-sectional plane similar to that of the echocardiographic (parasternal) long axis showing a common pattern of asymmetric LVH with wall thickening confined primarily to the AVS, which bulges prominently into the left ventricular outflow tract. LV denotes left ventricular free wall. B: A heart specimen in the transverse cross-sectional plane with a different pattern of hypertrophy; here, marked LV wall thickening is also asymmetric but predominantly in the PVS, whereas the AVS is only minimally thickened. C, D: Histology characteristic of the LV in HCM. C: The septal myocardium shows a markedly disordered architecture with adjacent hypertrophied cardiac muscle cells arranged at perpendicular and oblique angles. D: Bundles of hypertrophied cells show a disorganized, interwoven arrangement. E: An intramural coronary artery with an apparently narrowed lumen and thickened wall owing primarily to medial (M) hypertrophy. F: Extensive scarring of ventricular septum that is transmural in distribution, characteristic of the end-stage phase. LV, left ventricle; AVS, anterior ventricular septum; PVS, posterior ventricular septum; VS, ventricular septum. (Reproduced from Maron BJ, Bonow RO, Cannon RO 3rd, Leon MB, Epstein SE. Hypertrophic cardiomyopathy. Interrelations of clinical manifestations, pathophysiology, and therapy (1). N Engl J Med. 1987;316:780–789, with permission of Massachusetts Medical Society.) |
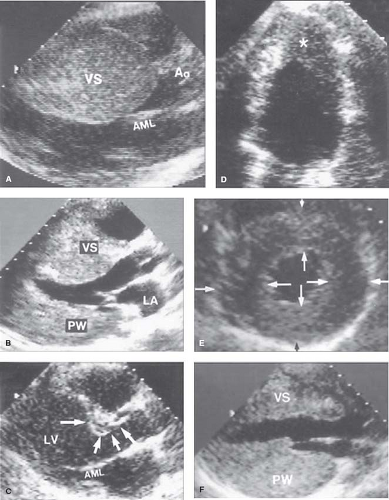 Figure 52.2 Patterns of LVH in HCM. Heterogeneous distribution and extent of LV wall thickening by echocardiography. A: Massive asymmetrical hypertrophy of VS with thickness >50 mm. B: Septal hypertrophy with distal portion considerably thicker than proximal region. C: Hypertrophy confined to proximal septum just below aortic valve (arrows). D: Hypertrophy localized to LV apex (asterisk)—that is, “apical HCM.” E: Relatively mild hypertrophy in symmetrical pattern showing similar or identical thicknesses within each segment (paired arrows). F: Inverted pattern with posterior free wall (PW) thicker (40 mm) than anterior VS. Calibration marks = 1 cm. Ao, aorta; AML, anterior mitral leaflet; LA, left atrium. (Reproduced from Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220–228, with permission of Elsevier.) |
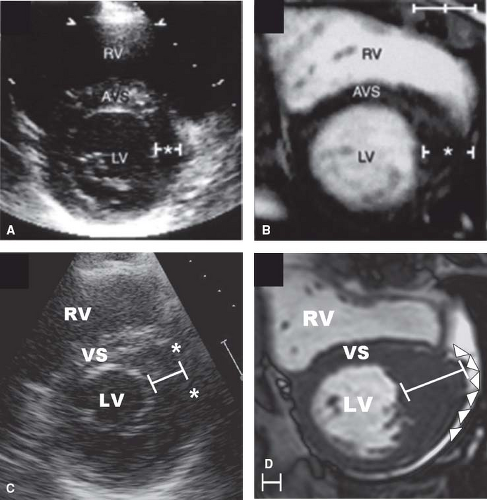 Figure 52.3 CMR identification of segmental LV wall thickening in HCM. Diagnosis. Two-dimensional echocardiogram (A) and comparative CMR (B) images acquired in short-axis plane at end diastole at the same level from 13-year-old asymptomatic identical twin. A: Echocardiogram shows normal anterolateral free wall thickness (asterisks). B: CMR shows segmental area of hypertrophy confined to anterolateral LV free wall (20 mm) and small portion of contiguous anterior septum. Calibration marks are 1 cm apart. (Reproduced from Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220–228, with permission of AHA). Echocardiogram (C) and comparative CMR (D) from 46-year-old man with HCM. C: Echocardiographic short axis shows anterolateral free wall thickness of 18 mm. D: CMR shows focal area of massive LVH (wall thickness, 35 mm) in the same region of LV, significantly underestimated by 2-D echocardiography. CMR finding defined high-risk status, prompting altered management strategy with an ICD recommendation for primary prevention of SD. Calibration markers are 1 cm apart. RV, right ventricle. (Reproduced from Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220–228, with permission of Elsevier.) |
Clinical diagnosis of HCM is usually made with 2-D echocardiography (6) or cardiovascular magnetic resonance (CMR) imaging (7). CMR may be advantageous by visualizing areas of segmental hypertrophy, specifically in the anterolateral free wall and LV apex, not always reliably identifiable with echocardiography (see Fig. 52.3) (7,28,29).
Hypertrophy may be diffuse and involve substantial portions of VS and LV free wall, including the most substantial hypertrophy observed in any cardiac disease, with wall thicknesses ranging up to 5 or 6 cm (6,7,41), often three to five times normal (see Fig. 52.2). However, notably, in about one-third of HCM patients, LVH is relatively mild (6,7) and confined to limited areas of the LV wall, usually the basal anterior septum (see Figs. 52.1 to 52.3) (6,7).
Segmental wall thickening confined to the LV apex (see Fig. 52.2), a form of HCM more commonly reported from Japan, produced a “spade” deformity of the distal LV and striking T-wave inversion and increased R-wave voltages in the lateral precordial leads on ECG (16,17,31). Notably, based on genotype–phenotype studies in HCM families, genetically affected relatives may have normal LV wall thicknesses (a subgroup referred to as “genotype positive/phenotype negative”) (32), and are considered at-risk for conversion to the HCM phenotype.
Serial echocardiographic investigations have shown that LVH in HCM is usually absent at birth, but commonly develops in a dynamic fashion after a period of prolonged latency (33,34,35,36). Typically, the HCM phenotype (i.e., LVH) is not complete until adulthood; during adolescence when body growth accelerates, genetically affected patients often show striking spontaneous increases in wall thickness (i.e., by an average of 100%) and more extensive distribution of hypertrophy (Fig. 52.4) (33). These evolving changes in LVH occurring during adolescence appear to be part of the genetically predetermined morphologic expression of HCM and are not per se associated with development or progression of symptoms or cardiac events. Mitral valve systolic anterior motion (SAM) and outflow obstruction may also develop in childhood, during the period of progressive LVH, and in the presence of a developmentally small outflow tract (35). Of note, in genetically affected children, 12-lead ECG abnormalities may be the initial clinical manifestation of the disease, even preceding appearance of LVH on 2-D echocardiogram or CMR (see Fig. 52.4) (34,36). On occasion, LVH has also been shown to develop or increase substantially well past childhood, in adult patients in their 30s to 50s (late-onset hypertrophy).
Mitral Valve
Structural abnormalities of the mitral valve, characteristic of many patients with LV outflow obstruction, may also be regarded as primary features of HCM (37,38). The majority of patients studied at necropsy (37) or by CMR (38) show important alterations in mitral valve size and shape. These primary abnormalities include increased overall mitral leaflet area (up to twice normal) owing largely to elongation of the leaflets (37) and, with considerable diversity, increased size of both anterior and posterior leaflets.
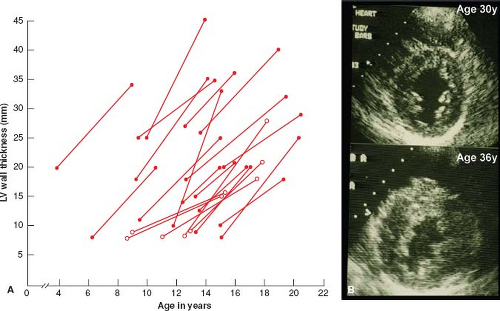 Figure 52.4 LV remodeling with development or progression of HCM phenotype. A: Childhood/adolescence. Increased echocardiographic LV wall thickness in patients with familial HCM, unassociated with changes in clinical course. Open symbols denote those without initial evidence of hypertrophy in any LV segment. B: Adulthood. Woman with myosin-binding protein C mutation shown at age 30 years with normal LV thickness (≤12 mm) in all segments of the wall (Top); 6 years later, at age 36, wall thickness has increased to 20 mm in both anterior ventricular septum and posterior septum, as well as anterolateral free wall. Bottom: LV wall thickness eventually increased to >30 mm, and the patient was prophylactically implanted with a defibrillator for primary prevention of SD. Calibration marks are 10 mm apart. VS, ventricular septum. (A: Reproduced from Maron BJ, Spirito P, Wesley Y, Arce J. Development and progression of left ventricular hypertrophy in children with hypertrophic cardiomyopathy. N Engl J Med. 1986;315:610–614. B: Reproduced from Maron BJ, Niimura H, Casey SA, et al. Development of left ventricular hypertrophy in adults with hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C mutations. J Am Coll Cardiol. 2001;38:315–321, with permission of Elsevier.) |
Secondary thickening of the anterior mitral leaflet may result from frequent contact with the VS, associated with localized endocardial thickening of the septum in the LV outflow tract adjacent to the anterior leaflet. Endocardial plaques on VS occur most commonly in the presence of LV outflow obstruction (due to mitral valve–septal systolic contact), but may also develop in nonobstructive HCM due to diastolic mitral valve–septal contact.
Histopathology
Cardiac muscle cells (myocytes) in VS and LV free wall have increased transverse diameter as well as bizarre shapes, often maintaining intercellular connections with several adjacent cells (39,41,42,43). Of note, many of these myocytes do not show normal parallel alignment, but are arranged in a chaotic, disorganized architecture, at oblique and perpendicular angles to each other; myofibrils within cardiac muscle cells may also appear disorganized. Cardiac muscle cell disarray is found in about 95% of patients dying of HCM, usually occupying substantial portions of LV myocardium, that is, about 33% of septum and 25% of LV free wall (39,43). Disorganized myocytes are not confined to markedly thickened segments of the LV wall but may also be present in nonhypertrophied areas, and with little relationship evident between the magnitude of wall thickening and extent of disarray (41). The finding of marked cellular disorganization in a few symptomatic infants with HCM suggests that this histologic abnormality can be present from birth (42).
It is likely that disorganized cells dispersed in the LV wall impair transmission of normal electrophysiologic impulses and predispose to disordered patterns and increased dispersion of electrical depolarization and repolarization. Therefore, this chaotic architecture may well serve as an electrically unstable arrhythmogenic substrate and the nidus for potentially lethal ventricular tachyarrhythmias and SD in HCM, or as a determinant of LV diastolic or systolic dysfunction.
Abnormalities of intramural coronary arteries (i.e., “small vessel disease”) are present in about 80% of patients studied at necropsy, most commonly in VS (see Fig. 52.1) (44,45). These vessels are characterized by thickening of the arterial wall with increased intimal or medial components and narrowing of the lumen. The abnormal arterioles may be responsible for silent myocardial ischemia and are most frequently observed within or in close proximity to areas of replacement fibrosis. HCM patients show variable severity and distribution of such fibrous tissue formation within LV myocardium, including patchy replacement fibrosis and grossly visible scars that may be extensive or even transmural, representing a repair process following myocyte death (see Fig. 52.1) (44,45,46,47,48,49). This myocardial fibrosis, long recognized at autopsy (48), can now be identified in vivo with contrast CMR (50). In addition, the interstitial (matrix) collagen compartment, constituting the structural framework of LV is substantially increased in size; its components (perimysial coils, pericellular weaves, and struts) are increased in number, appear morphologically abnormal, and often show disorganized arrangement (49).
Taken together, these findings suggest a form of small vessel disease in HCM that may be responsible for silent myocardial ischemia, necrosis, and ultimately replacement fibrosis (51,52,53,54,55). Areas of myocardial fibrosis and scarring can be responsible for LV systolic dysfunction (i.e., end-stage HCM) (56) and/or represent a substrate for clinically important ventricular tachyarrhythmias (57,58,59,60).
Pathophysiology
Left Ventricular Outflow Obstruction
The presence of LV outflow tract obstruction under basal conditions (gradient ≥30 mm Hg) over many years can be a determinant of progressive heart failure symptoms and cardiovascular death in HCM (Fig. 52.5) (61). The relationship of obstruction specifically to sudden cardiac death is much weaker, encumbered by particularly low positive predictive value (61). Obstruction in HCM represents true mechanical impedance to LV outflow, producing markedly increased intraventricular pressures, which can be detrimental to LV function by increasing myocardial wall stress and oxygen demand.
Dynamic subaortic obstruction in HCM is usually produced by the mitral valve, causing contact between the mitral valve and VS (SAM) (62). Characteristic of young patients with this disease is an abrupt anterior motion in which the elongated mitral valve leaflets move toward (or contact) the septum in early systole (with a 90-degree sharp-angled bend). Magnitude of the outflow gradient is directly related to duration of mitral–septal contact with prolonged contact throughout midsystole indicative of marked obstruction. Estimation of the magnitude of outflow obstruction is made conventionally with continuous wave Doppler (62), which obviates the need for diagnostic cardiac catheterization.
SAM and subaortic obstruction are determined by several geometric factors including the size of the outflow tract, distribution of LVH, and length of the mitral leaflets (37,63,64) as well as the ejection velocity. SAM appears to occur either by virtue of the high-velocity jet streaming through a narrowed outflow tract and pulling the mitral leaflets toward the septum (i.e., Venturi effect) or probably more likely due to drag (the hydrodynamic pushing force of flow) on the leaflets (65). The posteriorly directed mitral regurgitation jet, as a consequence of SAM, is usually only mild to moderate; severe mitral regurgitation in an HCM patient should raise the possibility of an intrinsic mitral valve abnormality such as myxomatous degeneration (66).
The subaortic gradient (and systolic ejection heart murmur) in HCM is characteristically dynamic (67) and may show considerable spontaneous variability, that is, reduced or abolished by interventions that decrease myocardial contractility (e.g., β-adrenergic receptor blocking drugs) or increase ventricular volume or arterial pressure (e.g., squatting, isometric handgrip, or phenylephrine administration). Alternatively, the outflow gradient and murmur can be augmented by circumstances that decrease arterial pressure or ventricular volume (e.g., the Valsalva maneuver or administration of nitroglycerin) or that increase contractility, such as premature ventricular contraction, standing, amyl nitrite inhalation, administration of isoproterenol, or exercise (2,3,4,5,67). Daily activities such as consuming a heavy meal may also transiently magnify the subaortic gradient (2,3). A large proportion of HCM patients without SAM or gradients at rest may nevertheless generate outflow obstruction with physiologic exercise. Indeed, fully 70% of HCM patients may have the propensity to develop obstruction (gradient ≥30 mm Hg) either at rest or with exercise (see Fig. 52.5) (23).
In infants and young children with HCM, obstruction to right ventricular outflow is not uncommon, usually occurring in association with subaortic obstruction. Subpulmonic gradients represent a form of fixed obstruction due to exaggerated hypertrophy of right ventricular musculature (i.e., crista supraventricularis muscle, papillary muscles, moderator band, or trabeculae) projecting into the relatively small outflow tract (68). The infrequency of subpulmonic obstruction in the adult HCM population suggests that these gradients probably disappear with body growth and cardiac remodeling.
Myocardial Ischemia
Regional myocardial ischemia (in the absence of atherosclerotic coronary artery disease) occurs commonly in HCM (51,52,53,54,69), as demonstrated by atrial pacing, reversible exercise-induced myocardial perfusion defects, dipyridamole stress ECG, necrosis, and the replacement fibrosis observed at autopsy or by CMR imaging (as delayed enhancement), or abnormalities recognized by positron emission tomography (PET) (54,69). Potential mechanisms by
which myocardial ischemia may occur in HCM include inadequate capillary density compared with greatly increased LV muscle mass, or alternatively, small vessel disease with narrowed intramural coronary arteries (44,45). It is difficult to clinically measure and quantitate the extent or location of myocardial ischemia in HCM patients, or consistently derive relevant correlations or prognostic information from these observations, although data from PET suggest ischemia is a determinant of progressive heart failure in HCM (69).
which myocardial ischemia may occur in HCM include inadequate capillary density compared with greatly increased LV muscle mass, or alternatively, small vessel disease with narrowed intramural coronary arteries (44,45). It is difficult to clinically measure and quantitate the extent or location of myocardial ischemia in HCM patients, or consistently derive relevant correlations or prognostic information from these observations, although data from PET suggest ischemia is a determinant of progressive heart failure in HCM (69).
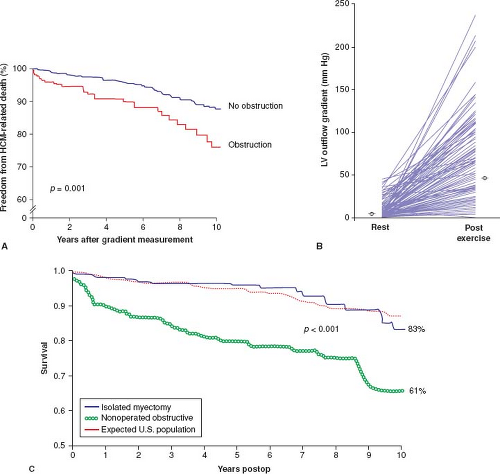 Figure 52.5 Left ventricular outflow tract obstruction. A: Probability of severe progressive heart failure (NYHA classes III or IV), heart failure death, or stroke in patients with LV outflow obstruction exceeds that in patients without obstruction (relative risk, 4.4; p <0.001). B: Changes in LV outflow tract gradient from resting (basal) conditions to immediately after termination of treadmill exercise. Each patient is depicted by a line connecting the two gradient measurements. θ indicates the mean. C: Following myectomy with relief of LV outflow obstruction, survival free from all-cause mortality compared with age- and gender-matched U.S. population and also nonoperated patients with obstruction (p <0.001). (A: Reproduced from Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295–303. B: Reproduced from Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114:2232–2239. C: Reproduced from Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:470–476.) |
Diastolic Dysfunction
Abnormalities of LV relaxation and filling have been identified by a variety of echocardiographic-Doppler methods in a majority of patients with HCM and which presumably contribute to (or are responsible for) symptoms of fatigue, exertional dyspnea, and angina pectoris (2,3,4,5,70,71,72,73,74,75,76), including end-stage heart failure in a small subset of patients with preserved systolic function. The rapid filling phase of diastole is significantly prolonged and associated with decreased
rate and volume compared with normal. Consequently, there is usually a compensatory increase in the contribution of atrial systole to overall LV filling. Conversely, diastolic dysfunction may be evident by echocardiographic parameters in the absence of both symptoms and outflow obstruction, unrelated to the severity or distribution of ventricular hypertrophy (70). Reduced ventricular distensibility probably results largely from those factors, which determine the passive elastic properties of the LV chamber, such as severity of hypertrophy, replacement and interstitial myocardial fibrosis, and myocyte architecture.
rate and volume compared with normal. Consequently, there is usually a compensatory increase in the contribution of atrial systole to overall LV filling. Conversely, diastolic dysfunction may be evident by echocardiographic parameters in the absence of both symptoms and outflow obstruction, unrelated to the severity or distribution of ventricular hypertrophy (70). Reduced ventricular distensibility probably results largely from those factors, which determine the passive elastic properties of the LV chamber, such as severity of hypertrophy, replacement and interstitial myocardial fibrosis, and myocyte architecture.
Genetics
HCM is a Mendelian trait with an autosomal dominant pattern of inheritance (Fig. 52.6) (1,2,3,4,5,10,19,20,77,78,79,80,81,82,83). Molecular studies with clinical genotype–phenotype correlations have provided important insights into the genetics of HCM, as well as access to laboratory-based diagnosis by virtue of detecting pathogenic sarcomere mutations, even in the absence of obvious clinical evidence of the disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


