52 Hypertrophic Cardiomyopathy
 Introduction
Introduction
By virtue of the broad variability in its phenotypic expression, hypertrophic cardiomyopathy (HCM) is a unique cardiovascular condition with a potential for the development of clinical symptoms during any phase of life, from infancy to greater than 90 years of age.1–7 The genetic foundation of HCM has been directly related to abnormalities of the genes encoding the cardiac sarcomere unit and may result in a complex disease phenotype that encompasses a spectrum of clinical and pathological presentations. In the past, the nomenclature regarding HCM has often been noted to be misleading. Idiopathic hypertrophic subaortic stenosis or hypertrophic obstructive cardiomyopathy (HOCM) typically described only a subset of patients with this disorder. With an improved understanding of the clinical heterogeneity of this process, hypertrophic cardiomyopathy appears to be a more appropriate descriptive term. The rapid demystification of the genetic underpinnings of HCM has greatly expanded our understanding of this entity. HCM is inherited in an autosomal dominant fashion with over 12 genes identified as being involved in the phenotypic manifestation.1,7–10 Three of those genes account for over 50% of the known cases of HCM.1,9,58 Traditionally, the diagnosis of HCM has been primarily clinical, involving the use of echocardiography to evaluate for certain characteristic features such as asymmetrical septal hypertrophy or systolic anterior motion of the mitral valve (SAM) with left ventricular outflow tract (LVOT) obstruction. While there have been dramatic advances in the understanding of the genetic predisposition for this disease state, the utility of genetic study for the absolute diagnosis remains preliminary at the present. However, the future holds promise that genetics will become a more reliable tool for establishing and confirming this diagnosis. The use of genotyping in risk stratification is also evolving.
 Epidemiology
Epidemiology
The prevalence of this genetic disorder is on the order of 1 : 500 in the general adult population and it is one of the more common cardiac genetic disorders known.1,6,9 While is not routinely accounted for in general practice, it is not uncommon to see these patients in tertiary referral centers. The clinical heterogeneity of this disorder plays into the difficulty in establishing a diagnosis. Often the presentation lacks the classic features noted on echocardiography. HCM is a disease process that is known to evolve with age, and the development of left ventricular hypertrophy has been observed to occur in children after full growth is attained.52,53,60 This can make diagnosis of HCM challenging and suggests that repeat evaluation in patients may be required to establish a diagnosis.
 Natural History of the Disease
Natural History of the Disease
The heterogeneity of HCM lies not only in its varied presentations but also in the natural history of disease in the patient population. Attempts to understand the links between genotype, phenotype, and natural history have as yet yielded only limited clinical associations. One of the most interesting aspects of the study of the natural history of patients with HCM is how selection bias has played a significant role in initial attempts to characterize patient outcomes. Earlier studies from tertiary referral centers implied ominously high annual mortality rates of 3% to 6%; however, this work was limited by a significant referral bias.1 More recent data from regional and community-based centers suggest an annual mortality of approximately 1%.3,4 However, in select populations, the annual mortality rate may be as high as 5% to 6%, particularly in those symptomatic patients who are eventually referred to larger centers.1,11,12 The clinical course of the HCM population is often difficult to predict and poses a challenge to clinicians. However, the options in terms of disease progression remain limited. The most feared and least predictable of the entities is sudden cardiac death, particularly in the younger population. More commonly, patients develop symptoms such as angina, syncope, or exertional dyspnea. These symptoms can become progressively worse over time, and such patients can progress toward end-stage heart failure with left ventricular failure. HCM patients also develop atrial fibrillation and are at risk for embolic strokes. A certain percentage of HCM patients remain asymptomatic and have a comparably normal life expectancy. However, at some point even they are at risk for the development of sudden cardiac death or atrial fibrillation. The challenge for clinicians is to closely follow those who eventually develop symptoms and to offer timely therapy when it is indicated.
 Clinical Presentation
Clinical Presentation
While the spectrum of clinical presentation in HCM is large, most patients are actually asymptomatic and diagnosed as the result of a murmur on exam, abnormal electrocardiogram (ECG), or unexplained left ventricular hypertrophy (LVH) discovered by echocardiography. The complex pathophysiological interplay between left ventricular outflow tract (LVOT) obstruction, diastolic dysfunction, myocardial ischemia, and mitral regurgitation generally results in the presenting complaints of exertional dyspnea, chest discomfort, syncope or near syncope, and sudden cardiac death. Symptomatic patients who will have an adverse clinical course will typically follow along one of several pathways: (1) those at high risk for sudden cardiac death; (2) progressive symptoms of exertional dyspnea and chest pain associated with presyncope or syncope in the setting of preserved LV function; (3) development of progressive congestive heart failure due to severe LV remodelling, resulting in systolic dysfunction; and (4) consequences of supraventricular or ventricular arrhythmias such as atrial fibrillation (AF) or ventricular tachycardia (VT).1,7,13–15 Sudden cardiac death is the most common presentation and source of mortality in HCM.1,7,14,16,17 In addition, sudden cardiac death (SCD) is the single leading cause of cardiovascular death among young people as well as the most common cause of mortality in competitive athletes.1,18 Most commonly observed in asymptomatic children and young adults, it appears that there is no advanced age at which the risk of SCD becomes negligible.19 While SCD is obviously the most fearsome and dramatic complication of HCM, those at high risk for SCD actually constitute only a small fraction of the disease spectrum,1,6,7,20,21 and much effort has been devoted to the premorbid identification of this subset of patients. Currently identified risk factors for SCD include prior cardiac arrest, family history of SCD, unexplained syncope or near syncope, left ventricular thickness greater than 30 mm, a high-risk genetic mutation (e.g., beta myosin heavy chain mutations Arg403Gln and Arg719Gln), hypotensive response during exercise stress testing, and nonsustained VT on Holter monitoring (Table 52-1).1,7,14,20,22–27 In addition, an LVOT gradient greater than 30 mm Hg has been associated with an increased risk of SCD, progression to heart failure, and morbidity related to arrhythmia, including stroke.28,29 However, an incremental increase in the subaortic gradient above 30 mm Hg has not been demonstrated to impart any additional risk. It is uncommon for HCM patients to suffer SCD without at least one of the aforementioned risk factors (<3%).20 It has been suggested that the etiology of SCD in this population is related to the development of complex ventricular tachyarrhythmia,7,30,31 often during mild to moderate physical exertion and with a circadian predilection for the early morning hours.32 Chest pain, both typical and atypical in character, is a common feature in HCM and has been reported in up to 80% of patients in this population.33 In many cases, angiography reveals normal coronary arteries. Despite this finding, numerous studies incorporating nuclear single photon emission computed tomography (SPECT), positron emission tomography (PET), and magnetic resonance imaging (MRI) technologies have demonstrated significant reversible and nonreversible myocardial perfusion defects in this subset of patients, including autopsy data reporting findings of myocardial infarction in up to 15% of such patients.1,7,33–37 Collectively, these data have led to a mounting body of evidence suggesting that microvascular dysfunction may have a pivotal role in the development of myocardial ischemia and infarction in this group. The etiology of microvascular dysfunction is probably multifactorial and due in part to arteriolar medial hypertrophy, resulting in reduced luminal diameter, impaired coronary vasodilatory response, and a supply : demand mismatch due to an abnormally thickened ventricle.1,7,34,38 In addition, early work has suggested that evidence of microvascular dysfunction, as demonstrated by PET, is an independent predictor of increased mortality and may portend a worse prognosis years prior to the development of clinical deterioration.39 Syncope in patients with HCM is not an uncommon phenomenon and has a diverse array of possible etiologies, making the exact determination of mechanism challenging. While regarded as an ominous prognostic sign and known risk factor for SCD in the younger population, syncope in the adult population has not been independently associated with premature demise, and recurrent episodes are rarely reported in patients who have suffered SCD.40–42 Arrhythmic sources of syncope may be supraventricular, such as atrial fibrillation or flutter, or ventricular, such as ventricular tachycardia or fibrillation. Hemodynamic mechanisms of syncope all result in a sudden and severe reduction in cardiac output that may involve ischemia, outflow tract obstruction, or severe diastolic dysfunction. Additionally, it has been suggested that activation of left ventricular baroreceptors due to elevated intracavity pressures may induce reflex hypotension and a consequent syncopal episode in a select subgroup of patients.43 Heart failure—as manifested by a symptom complex of exertional dyspnea, orthopnea, and progressive fatigue—is most commonly encountered in adult patients with HCM, but it has been described in the juvenile population as well.1 Usually, in the setting of preserved systolic function, symptoms are most commonly the consequence of diastolic dysfunction due to an abnormally thickened and noncompliant ventricle.7 The combined influence of other variables such as ischemia, AF, and mitral regurgitation may also play a significant role in the development of hemodynamic decompensation in this population. A smaller number of patients with HCM and heart failure may have significantly reduced left ventricular systolic function and chamber enlargement. It is important to recognize this subset of patients, given the potential alteration in therapeutic strategy.41 Atrial fibrillation complicates the course of approximately 20% of patients with HCM and is associated with an increased risk of heart failure-related death.17,24 The risk seems to be substantially greater in the subset of patients with outflow tract obstruction or an earlier onset of arrhythmia (<50 years of age). Advancing age, left atrial enlargement, and congestive symptoms are independently linked with the development of atrial fibrillation. While strongly associated with an increased risk of fatal and nonfatal stroke, atrial fibrillation does not appear to be a risk factor for the development of SCD, and approximately one-third of patients have no long-term sequelae from this arrhythmia.24 Severe functional deterioration due to dyspnea, chest pain, palpitations, or pulmonary edema may complicate the course of the chronically affected. This is most likely due to the loss of atrial contraction, reduction in diastolic filling time, and exacerbation of underlying ischemia.7,24 The nature of the clinical presentation may also be affected by a particular patient’s age or gender. In contrast to their younger counterparts, elderly patients with HCM often develop marked symptomatology at an advanced age (>55 years), have lesser degrees of left ventricular hypertrophy usually confined to the septum, and a dynamic subaortic gradient due to restricted excursion of the often anteriorly displaced mitral leaflets and posteriorly directed septal motion.44 While HCM seems to have a male predominance, female patients often present at a later age, are more symptomatic, and are at a greater risk of death due to heart failure or stroke.45
TABLE 52-1 Risk Factors for Sudden Cardiac Death
LVOT, left ventricular outflow tract; SCD, sudden cardiac death; VT, ventricular tachycardia.
 Diagnosis
Diagnosis
Echocardiography
Given its safety and ubiquity, two-dimensional echocardiography is the most common method for establishing the clinical diagnosis of HCM via the identification of a thickened, nondilated left ventricle in the absence of comorbidities known to cause such a degree of left ventricular hypertrophy (i.e., hypertension or aortic stenosis).1,7,46 Classically thought to involve primarily the ventricular septum, the morphological expression of left ventricular hypertrophy is extremely heterogeneous and virtually any pattern of thickening may be observed.1,7,47 In addition, there are significant differences in the pattern of hypertrophy between young and elderly patients. Elderly patients are often found to have an elliptical ventricular cavity with hypertrophy predominantly of the basal septum. In contrast, young patients (<55 years) often have a crescent-shaped ventricular cavity associated with diffuse hypertrophy of the interventricular septum.48 While a maximal wall thickness greater than 15 mm is the traditional echo cardiographic benchmark for HCM, the degree of hypertrophy may demonstrate considerable variability (with a mean thickness of approximately 22 mm).47 It is important to realize, however, that the paucity of characteristic LVH (>15 mm) on echocardiographic exam does not exclude the presence of a HCM gene mutation.7,9,49–51 Thus, serial echocardiographic assessment may be necessary for adequate identification of suspected carriers, especially in the younger population, in whom the development of LVH may be delayed until after puberty.1,14,51
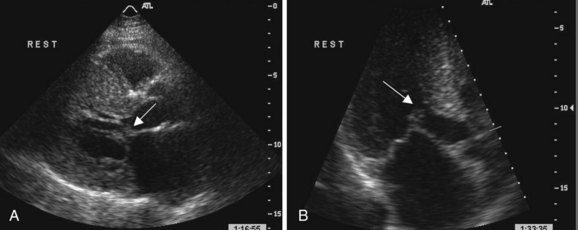
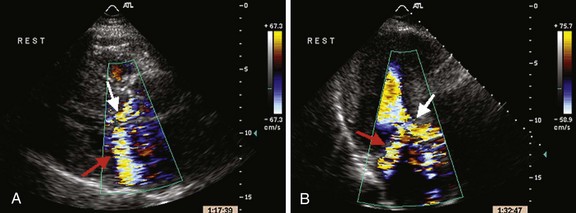
LVOT is observed in approximately 20% of patients with HCM and is usually dynamic in nature.17,28 Subaortic obstruction is due to systolic anterior motion (SAM) of the anterior mitral leaflet, resulting in mitral-septal contact during midsystole7 (Fig. 52-1). Obstruction may not be present under resting conditions but can be provoked by pharmacological (i.e., amyl nitrite) or physiological maneuvers (i.e., Valsalva). Significant mitral regurgitation frequently accompanies SAM owing to distortion of the valvular apparatus and malcoaptation of the anterior and posterior leaflets (Fig. 52-2). Mitral regurgitation is also observed in up to 30% of patients who do not demonstrate obstructive physiology primarily due to leaflet prolapse, chordal rupture, or trauma resulting in calcification or fibrosis.52 Less commonly, a midcavity gradient is formed because of the anomalous insertion of the anterolateral papillary muscle directly onto the anterior mitral leaflet or an exaggerated proliferation of midventricular papillary musculature coming into apposition with the ventricular septum.7,53,54 While the threshold for therapeutic intervention has traditionally been a gradient >50 mm Hg, it has been demonstrated that the presence of a resting LVOT obstruction >30 mm Hg is an independent predictor of death from heart failure or stroke, progression of heart failure symptoms, and reduced functional capacity as well as SCD.28 It is important not to misinterpret the Doppler spectral display of mitral regurgitation for LVOT gradient given its frequent presence in the setting of obstruction and its close spatial orientation to the LVOT. In the setting of SAM, mitral regurgitation is usually posteriorly directed into the left atrium and is often difficult to distinguish from LVOT flow. It is most useful to sweep anterior to posterior with continuous Doppler to distinguish these two flows. Given the magnitude of left ventricular hypertrophy consummate with HCM, it is not surprising that more than 80% of patients have evidence of diastolic dysfunction by echocardiogram. This is manifested by reduced maximal flow velocity in early diastole, an increase in isovolumic relaxation time, and increased atrial contribution to ventricular filling.46,55 These findings are similar in patients both with and without an LVOT gradient or cardiac symptoms, suggesting that diastolic dysfunction may be an earlier clinical manifestation in the spectrum of this disease process. Several studies have suggested that the presence of significant diastolic dysfunction by transthoracic or tissue Doppler echocardiography may imply an increased risk of cardiac arrest, VT, or progression to significant cardiac symptoms.56
Electrocardiography
ECG findings in HCM are extremely heterogeneous and the vast majority (>90%) of patients will have demonstrable abnormalities.1,7,57,58 However, no pattern is highly specific for the condition and the presence of a normal tracing does not imply absence of the disease state.58,59 Increased voltages consistent with LVH and early repolarization abnormalities are most commonly encountered, while left axis deviation, left atrial enlargement, T-wave inversion, and nonspecific ST-segment abnormalities are also frequently noted. The degree of LVH by ECG does not appear to correlate with the magnitude of hypertrophy when assessed by echocardiography.60 In a subset of Japanese patients with hypertrophy primarily limited to the ventricular apex, giant T-wave inversions are frequently noted in the anterior leads; these are often termed Yamaguchi’s disease.61 Pathological Q waves, often in the inferolateral leads, may be observed in up to 50% of patients with known HCM. While not apparent on the surface ECG, approximately one-third of patients have delayed His-Purkinje conduction on formal electrophysiological studies, possibly owing to strain on the anterior fasciculus, which overlies the hypertrophied ventricle.57
Magnetic Resonance Imaging
In comparison with traditional echocardiography, cardiac magnetic resonance imaging (CMRI) offers the advantages of superior resolution with precise morphological characterization, enhanced tissue contrast capability, and production of three-dimensional images.62 These advantages result in the ability of CMRI to better detect areas of hypertrophy that are not well visualized or missed by traditional echocardiography. Particularly in patients with atypical hypertrophy of the anterolateral free wall, CMRI is a powerful adjunctive tool in the diagnosis of HCM.62
Through delayed hyperenhancement techniques, CMRI has demonstrated that asymptomatic patients with HCM frequently have patchy foci of myocardial scarring at the junction of the interventricular septum and the right ventricular free wall. Furthermore, scarring was limited to the areas of abnormal hypertrophy and the degree of scarring was proportional to the magnitude of hypertrophy, while wall thickening was inversely related.35 In addition, a greater extent of hyperenhancement has been positively associated with patients at high risk for SCD and in those with progressive disease.63 CMRI also allows for better characterization of papillary muscle insertion and orientation. It is not uncommon to see hypertrophic, displaced, or distorted papillary muscles contributing to the obstruction and/or mitral valve dysfunction. Considering all these, CMRI is a valuable adjunctive imaging modality for the diagnosis of HCM.
Catheterization and Hemodynamics
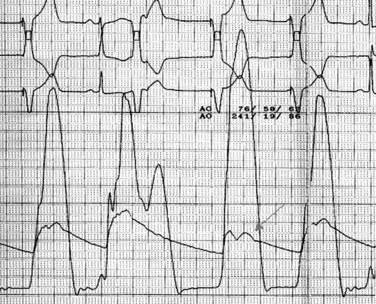
Given the wealth of hemodynamic and anatomical data that can be derived noninvasively by echocardiography, cardiac catheterization is not required for the diagnosis of HCM. Catheterization is often employed, however, if noninvasive imaging is of insufficient quality to quantify the degree or location of obstruction, to evaluate for coronary disease prior to a planned surgical therapy (i.e., myectomy or pacemaker), or if anginal symptoms that may be attributable to ischemia are present in older patients. The coronary arteries in patients with HCM are usually normal and typically of large caliber. Quite different from intramyocardial “bridging,” compression of the left anterior descending artery (LAD) may be observed during systole due to contraction of the hypertrophied ventricle, resulting in a “sawfish” appearance.64 Ventriculography may demonstrate systolic cavity obliteration, varying degrees of mitral regurgitation, and occasionally the hypertrophied septum prolapsing into the LVOT. Direct measurement and localization of the gradient is easily obtained by passing a multipurpose catheter into the apical portion of the left ventricle and slowly withdrawing it while continuously monitoring the pressure waveform. Use of a wire via a guide catheter often results in increased control during the pullback and a more accurate determination of the level of obstruction. As opposed to what is observed in aortic stenosis (AS), the gradient is reduced prior to crossing the aortic valve. This same technique can be performed using simultaneous aortic and LV pressure waveforms to allow side-by-side comparison. The gradient in HCM is characteristically labile and various pharmacological and physiological maneuvers similar to echocardiography may be employed to accentuate the obstruction while in the catheterization laboratory. The term postextrasystolic potentiation64a refers to the augmentation of LV pressure with a concomitant decrement in the aortic systolic and pulse pressures as a result of increased LVOT obstruction in the cardiac cycle that follows a premature ventricular contraction (PVC). Postextrasystolic increase in gradient between LV and aorta is seen even with aortic stenosis but, unlike the case in HCM, pulse pressure (stroke volume) does not decrease. This is because in aortic stenosis, the larger stroke volume of the postextrasystolic beat leads to a higher gradient with no change in the severity of obstruction (Fig. 52-3).
Genetic Overview
HCM is the result of mutations to genes primarily encoding sarcomeric proteins that regulate contractile, regulatory, and structural functions; they are inherited in an autosomal dominant manner.1,7–10 To date, more than 400 mutations have been described involving 12 genes, the most common of which include cardiac troponins T, C, and I, cardiac myosin-binding protein C, cardiac beta- and alpha-myosin heavy chains, myosin ventricular essential and regulatory light chains, cardiac alpha actin, and titin.8,10 While most of these mutations are missense with resultant substitution of the correct amino acid for another, deletions, insertions, and splice-site mutations are also well described.65 Several nonsarcomeric mutations that produce phenotypes similar to HCM have been identified. PRKAG2 affects the regulatory subunit of the AMP-activated protein kinase and may result in preexcitation, progressive conduction system abnormalities, and mild ventricular hypertrophy due to aberrant accumulation of glycogen within the myocyte.65–67 Mutations of 2 alpha-galactosidase or acid alpha-1, 4-glucosidase (both lysosome-associated membrane proteins) frequently result in multisystem glycogen storage disease and may also present with extreme LVH associated with ventricular preexcitation and mental retardation.65,66,68 There is great phenotypical heterogeneity among carriers of the same mutations, in part due to the effect of modifier genes and environmental factors.7,69 While it has been known that many young carriers may not demonstrate the morphological characteristics of the disease state until after adolescence, it has now been demonstrated that phenotypical expression of LVH can be delayed into late adulthood owing to incomplete penetrance of mutations involving cardiac myosin-binding protein C or troponin T.7,49,50,59,70 While the majority of studied HCM cases involve familial mutations, sporadic cases are also well described and may constitute a significant proportion of the population. Recent work involving the systematic molecular screening of known HCM cases has demonstrated that two mutations (MYBPC3 and MYH7) may account for 82% of familial cases and that mutations were detected in up to 60% of “sporadic cases.”10 These data would imply that a relatively limited screening process may be sufficient to identify the culprit gene in most familial cases and that identifiable mutations are responsible for the majority of sporadic cases. Given the fact that a number of studies have identified specific genetic mutations (Table 52-2) seemingly associated with a worse clinical prognosis and higher rates of SCD, there has been initial enthusiasm that genetic testing could prospectively identify patients at higher risk for premature demise.1,9,14,27,49,50,71 However, significant limitations including selection bias, the small number of included familial cohorts, low frequency of specific gene mutations, and variability of the phenotypical product have hindered most of these genotype-phenotype correlation studies.71,72 Thus, because of the numerous genetic and environmental influences affecting the phenotypical product, there remains a great deal of clinical heterogeneity associated with specific mutations, making accurate risk stratification based on genetic analysis alone impractical at this time.
TABLE 52-2 Sarcomeric Gene Mutations of Hypertrophic Cardiomyopathy
 Treatment
Treatment
Medical Therapy
Medical therapy should be considered the initial therapeutic approach for the treatment of symptoms arising from the numerous pathophysiological processes constituting HCM. Because of the relatively small number of cases, pharmacological therapy for HCM is largely based on expert opinion, clinical experience, and retrospective observational analyses. While patients with LVOT obstruction make up the greatest proportion of the symptomatic population, a significant number of nonobstructing patients may also suffer the consequences of diastolic dysfunction, such as heart failure, angina, and atrial fibrillation.1,7,14 Given the increasing utilization of early genetic and echocardiographic screening of athletes and affected families, it has become apparent that a significant percentage of phenotypically affected patients are entirely asymptomatic for an extended time and, while somewhat controversial, available data suggest that this population does not warrant empirical therapy until and if symptoms develop.1,5,7,17 Historically, the pharmacological treatment of HCM has been limited to beta blockers, verapamil, and disopyramide.



