Jeffrey I. Weitz
Hemostasis, Thrombosis, Fibrinolysis, and Cardiovascular Disease
Hemostasis preserves vascular integrity by balancing the physiologic processes that maintain blood fluidity under normal circumstances and preventing excessive bleeding after vascular injury. Preservation of blood fluidity depends on an intact vascular endothelium and a complex series of regulatory pathways that maintain platelets in a quiescent state and keep the coagulation system in check. In contrast, arrest of bleeding requires rapid formation of hemostatic plugs at sites of vascular injury to prevent exsanguination. Perturbation of hemostasis can lead to thrombosis, which can occur in arteries or veins and causes considerable morbidity and mortality. Arterial thrombosis is the most common cause of acute coronary syndromes, ischemic stroke, and limb gangrene, whereas thrombosis in the deep veins of the leg leads to post syndrome and pulmonary embolism, which can be fatal (see also Chapter 73).
Most arterial thrombi form on top of disrupted atherosclerotic plaque because plaque rupture exposes thrombogenic material in the plaque core to blood (see also Chapter 41). This material then triggers platelet aggregation and fibrin formation, which results in the generation of a platelet-rich thrombus that temporarily or permanently occludes blood flow.1 Transient occlusion of blood flow in coronary arteries may trigger unstable angina, whereas persistent obstruction causes myocardial infarction. The same processes can occur in the cerebral circulation, where temporary arterial occlusion may be manifested as a transient ischemic attack and permanent occlusion can lead to a stroke.
In contrast to arterial thrombi, venous thrombi rarely form at sites of obvious vascular disruption.2 Although venous thrombi can develop after surgical trauma to veins or arise secondary to indwelling venous catheters, they usually originate in valve cusps of the deep veins of the calf or in muscular sinuses, where there is stasis. Sluggish blood flow in these veins reduces oxygen supply to the avascular valve cusps. Hypoxemia induces endothelial cells lining the valve cusps to express adhesion molecules, which tether tissue factor–bearing leukocytes and microparticles onto their surface. Tissue factor–bearing leukocytes and microparticles adhere to these activated cells and induce coagulation.3 In addition, webs of DNA released from activated neutrophils, so-called neutrophil extracellular traps (NETs), also contribute to thrombosis by providing a scaffold that binds platelets and promotes their activation and aggregation.4 Impaired blood flow exacerbates local thrombus formation by reducing clearance of activated clotting factors. Calf vein thrombi that extend into the proximal veins of the leg can dislodge and travel to the lungs to produce pulmonary embolism.
Arterial and venous thrombi contain platelets and fibrin, but the proportions differ. Arterial thrombi are rich in platelets because of high shear in the injured arteries. In contrast, venous thrombi, which form under low-shear conditions, contain relatively few platelets and consist mostly of fibrin and trapped red cells.2 Because of the predominance of platelets, arterial thrombi appear white, whereas venous thrombi appear red as a result of the trapped red cells.
The antithrombotic drugs used for prevention and treatment of thrombosis target components of thrombi and include antiplatelet drugs, which inhibit platelets; anticoagulants, which attenuate coagulation; and fibrinolytic agents, which induce fibrin degradation (Fig. 82-1). With the predominance of platelets in arterial thrombi, strategies to inhibit or treat arterial thrombosis focus mainly on antiplatelet agents, although in the acute setting, anticoagulants and fibrinolytic agents may also be used. When arterial thrombi are occlusive and require rapid restoration of blood flow, mechanical and/or pharmacologic methods enable thrombus extraction, compression, or degradation. Although rarely used for this indication, warfarin prevents recurrent ischemic events after acute myocardial infarction. The recent observation that the addition of low-dose rivaroxaban, an oral factor Xa inhibitor, to dual-antiplatelet therapy reduces recurrent ischemic events and stent thrombosis in patients with acute coronary syndrome highlights the potential usefulness of anticoagulants for secondary prevention (see also Chapters 52 and 53).
Anticoagulants are the mainstay for prevention and treatment of venous thromboembolism (VTE) because venous thrombi contain predominantly fibrin.2 Antiplatelet drugs are less effective than anticoagulants for prevention of venous thrombosis because of the limited platelet content of venous thrombi. Nonetheless, when given for secondary prevention, aspirin produces about a 30% reduction in risk for recurrent VTE,5,6 a finding that highlights the overlap between venous and arterial thrombosis. Selected patients with VTE benefit from fibrinolytic therapy6—for example, patients with massive or submassive pulmonary embolism achieve more rapid restoration of pulmonary blood flow with systemic or catheter-directed fibrinolytic therapy than with anticoagulant therapy alone (see Chapter 73). Similarly, some patients with extensive deep vein thrombosis in the iliac and/or femoral veins may have a better outcome with catheter-directed fibrinolytic therapy and/or mechanical thrombus extraction in addition to anticoagulants.
Starting with a review of hemostasis and thrombosis that highlights the processes involved in platelet activation and aggregation, blood coagulation, and fibrinolysis, this chapter focuses on antiplatelet, anticoagulant, and fibrinolytic drugs in common use. It also provides a brief overview of new antithrombotic drugs in advanced stages of development.
Hemostatic System
The major components of the hemostatic system are the vascular endothelium, platelets, and coagulation and fibrinolytic systems.
Vascular Endothelium (See Chapter 41)
A monolayer of endothelial cells lines the intimal surface of the circulatory tree and separates blood from the prothrombotic subendothelial components of the vessel wall. Accordingly, the vascular endothelium encompasses about 1013 cells and covers a vast surface area. Rather than serving as a static barrier, healthy vascular endothelium is a dynamic organ that actively regulates hemostasis by inhibiting platelets, suppressing coagulation, and promoting fibrinolysis.
Platelet Inhibition
Endothelial cells synthesize prostacyclin and nitric oxide and release them into blood. These agents not only serve as potent vasodilators but also inhibit platelet activation and subsequent aggregation by stimulating adenylate cyclase and increasing intracellular levels of cyclic adenosine monophosphate (cAMP). In addition, endothelial cells express CD39 on their surface. This membrane-associated ecto-adenosine diphosphatase (ecto-ADPase) attenuates platelet activation by degrading ADP.7
Anticoagulant Activity
Intact endothelial cells play an essential part in the regulation of thrombin generation. Endothelial cells express heparan sulfate proteoglycans on their surface. Like medicinal heparin, heparan sulfate binds circulating antithrombin and enhances its activity. Heparan sulfate proteoglycans also bind tissue factor pathway inhibitor (TFPI), a naturally occurring inhibitor of coagulation.8 Administration of heparin or low-molecular-weight heparin (LMWH) displaces glycosaminoglycan-bound TFPI from the vascular endothelium, and the TFPI released may contribute to the antithrombotic activity of these drugs by inhibiting tissue factor–bound factor VIIa in a factor Xa–dependent fashion.
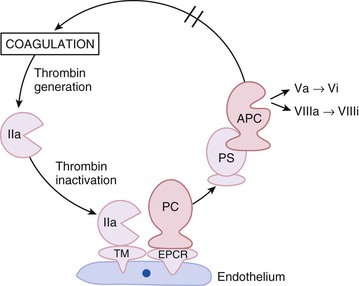
Endothelial cells are key components of the protein C anticoagulant pathway because they express thrombomodulin and endothelial cell protein C receptor (EPCR) on their surface.9 The protein C pathway is initiated when thrombin binds to thrombomodulin. Once bound, the substrate specificity of thrombin is altered such that it no longer acts as a procoagulant but becomes a potent activator of protein C (Fig. 82-2). Activated protein C serves as an anticoagulant by degrading and inactivating activated factor V and factor VIII (factors Va and VIIIa, respectively), key cofactors involved in thrombin generation. The EPCR on the endothelial cell surface enhances this pathway by binding protein C and presenting it to the thrombin-thrombomodulin complex for activation.9
Fibrinolytic Activity
The vascular endothelium promotes fibrinolysis by synthesizing and releasing tissue and urokinase plasminogen activator (t-PA and u-PA, respectively), which initiate fibrinolysis by converting plasminogen to plasmin.10 Endothelial cells in most vascular beds synthesize t-PA constitutively. In contrast, perturbed endothelial cells produce u-PA in the settings of inflammation and wound repair.
Endothelial cells also produce type 1 plasminogen activator inhibitor (PAI-1), the major regulator of both t-PA and u-PA. Therefore net fibrinolytic activity depends on the dynamic balance between release of plasminogen activators and PAI-1. Fibrinolysis is localized to the endothelial cell surface because these cells express annexin II, a coreceptor for plasminogen and t-PA that promotes their interaction. Hence healthy vessels actively resist thrombosis and help maintain platelets in a quiescent state.10
Platelets
Platelets are anucleate particles released into the circulation after fragmentation of bone marrow megakaryocytes. Because they are anucleate, platelets have limited capacity to synthesize proteins. Thrombopoietin, a glycoprotein synthesized in the liver and kidneys, regulates megakaryocytic proliferation and maturation, as well as platelet production.11 Once they enter the circulation, platelets have a life span of 7 to 10 days.
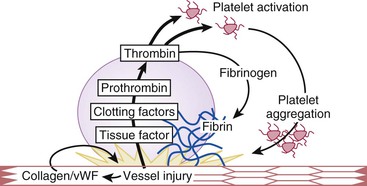
Damage to the intimal lining of the vessel exposes the underlying subendothelial matrix. Platelets home to sites of vascular disruption and adhere to the exposed matrix proteins. Adherent platelets undergo activation and not only release substances that recruit additional platelets to the site of injury but also promote thrombin generation and subsequent fibrin formation (Fig. 82-3). A potent platelet agonist, thrombin amplifies platelet recruitment and activation. Activated platelets then aggregate to form a plug that seals the leak in the vasculature. An understanding of the steps in these highly integrated processes helps pinpoint the sites of action of antiplatelet drugs and rationalizes the usefulness of anticoagulants for the treatment of arterial and venous thrombosis.
Adhesion
Platelets adhere to exposed collagen and von Willebrand factor (vWF) and form a monolayer that supports and promotes thrombin generation and subsequent fibrin formation.12 These events depend on constitutively expressed receptors on the platelet surface, α2β1 and glycoprotein VI (GP VI), which bind collagen, and GP Ibα and GP IIb/IIIa (αIIbβ3), which bind vWF. The platelet surface is crowded with receptors, but those involved in adhesion are the most abundant; every platelet has approximately 80,000 copies of GP IIb/IIIa and 25,000 copies of GP Ibα. Receptors cluster in cholesterol-enriched subdomains, which renders them more mobile, thereby increasing the efficiency of platelet adhesion and subsequent activation.13
Under low-shear conditions, collagen can capture and activate platelets on its own. The captured platelets undergo cytoskeletal reorganization, which causes them to flatten out and adhere more closely to the damaged vessel wall. Under high-shear conditions, however, collagen and vWF must act in concert to support optimal platelet adhesion and activation. The vWF synthesized by endothelial cells and megakaryocytes assembles into multimers that range from 550 to greater than 10,000 kDa.14 When released from storage in the Weibel-Palade bodies of endothelial cells or the alpha granules of platelets, most of the vWF enters the circulation, but the vWF released from the abluminal surface of endothelial cells accumulates in the subendothelial matrix, where it binds collagen via its A3 domain. This surface-immobilized vWF can simultaneously bind platelets via its A1 domain. In contrast, circulating vWF does not react with unstimulated platelets. This difference in reactivity probably reflects the conformation of vWF; circulating vWF is in a coiled conformation, which prevents access of its platelet binding domain to vWF receptors on the platelet surface, whereas immobilized vWF assumes an elongated shape, which exposes the A1 domain. In their extended conformation, large vWF multimers act as the molecular glue that tethers platelets to the damaged vessel wall with sufficient strength to withstand higher shear force. Large vWF multimers provide additional binding sites for collagen and heighten platelet adhesion because platelets have more vWF receptors than collagen receptors.15 Adhesion to collagen or vWF results in platelet activation, the next step in platelet plug formation.
Activation
Adhesion to collagen and vWF initiates signaling pathways that result in platelet activation. These pathways induce cyclooxygenase-1 (COX-1)-dependent synthesis and release of thromboxane A2 and trigger the release of ADP from storage granules. Thromboxane A2 is a potent vasoconstrictor and, like ADP, locally activates ambient platelets and recruits them to the site of injury, thereby expanding the platelet plug. To activate platelets, thromboxane A2 and ADP must bind to their respective receptors on the platelet membrane. The thromboxane receptor (TP) is a G protein–coupled receptor that is found on platelets and the endothelium, which explains why thromboxane A2 induces vasoconstriction as well as platelet activation.16 ADP interacts with a family of G protein–coupled receptors on the platelet membrane.17,18 The most important of these is P2Y12, which is the target of the thienopyridines and ticagrelor, but P2Y1 also contributes to ADP-induced platelet activation, and maximal ADP-induced platelet activation requires activation of both receptors. A third ADP receptor, P2X1, is an adenosine triphosphate (ATP)-gated calcium channel. Platelet storage granules contain ATP, as well as ADP; the ATP released during the platelet activation process may contribute to the platelet recruitment process in a P2X1-dependent fashion.
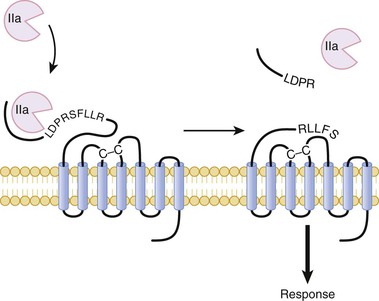
Although TP and the various ADP receptors signal through different pathways, they all trigger an increase in the intracellular concentration of calcium in platelets. The increase in calcium induces changes in platelet shape via cytoskeletal rearrangement, granule mobilization and release, and subsequent platelet aggregation. Activated platelets promote coagulation by expressing phosphatidylserine on their surface, an anionic phospholipid that supports the assembly of coagulation factor complexes. Once assembled, these clotting factor complexes trigger a burst of thrombin generation and subsequent fibrin formation. In addition to converting fibrinogen to fibrin, thrombin amplifies platelet recruitment and activation and promotes expansion of the platelet plug. Thrombin binds to protease-activated receptor types 1 and 4 (PAR-1 and PAR-4, respectively) on the platelet surface and cleaves their extended amino-terminals (Fig. 82-4), thereby generating new amino-terminals that serve as tethered ligands that bind and activate the receptors.19 Low concentrations of thrombin cleave PAR-1, whereas PAR-4 cleavage requires higher thrombin concentrations. Cleavage of either receptor triggers platelet activation.
In addition to providing a surface on which clotting factors assemble, activated platelets also promote fibrin formation and subsequent stabilization by releasing factor V, factor XI, fibrinogen, and factor XIII. Thus there is coordinated activation of platelets and coagulation, and the fibrin network that results from the action of thrombin helps anchor the platelet aggregates at the site of injury. Activated platelets also release adhesive proteins, such as vWF, thrombospondin, and fibronectin, which may augment platelet adhesion at sites of injury, as well as growth factors such as platelet-derived growth factor (PDGF) and transforming growth factor-beta (TGF-β), which promote wound healing. Platelet aggregation is the final step in formation of the platelet plug.
Aggregation
Platelet aggregation links platelets to each other to form clumps. GP IIb/IIIa mediates these platelet-to-platelet linkages. On nonactivated platelets, GP IIb/IIIa exhibits minimal affinity for its ligands. On platelet activation, GP IIb/IIIa undergoes a conformational change that reflects transmission of inside-out signals from its cytoplasmic domain to its extracellular domain.18 This transformation enhances the affinity of GP IIb/IIIa for its ligands, fibrinogen, and under high-shear conditions, vWF. Arg-Gly-Asp (RGD) sequences located on fibrinogen and vWF, as well as a platelet-binding Lys-Gly-Asp (KGD) sequence on fibrinogen, mediate their interaction with GP IIb/IIIa. When subjected to high shear, circulating vWF elongates and exposes its platelet binding domain, which enables its interaction with the conformationally activated GP IIb/IIIa.15 Divalent fibrinogen and multivalent vWF molecules serve as bridges and bind adjacent platelets together. Once bound to GP IIb/IIIa, fibrinogen and vWF induce outside-inside signals that augment platelet activation and result in the activation of additional GP IIb/IIIa receptors, thus creating a positive feedback loop. Because GP IIb/IIIa acts as the final effector in platelet aggregation, it is a logical target for potent antiplatelet drugs. Fibrin, the ultimate product of the coagulation system, tethers the platelet aggregates together and anchors them to the site of injury.
Coagulation
Coagulation results in the generation of thrombin, which converts soluble fibrinogen to fibrin. Coagulation occurs through the action of discrete enzyme complexes composed of a vitamin K–dependent enzyme and a nonenzyme cofactor that assemble on anionic phospholipid membranes in a calcium-dependent fashion. Each enzyme complex activates a vitamin K–dependent substrate that becomes the enzyme component of the subsequent complex (Fig. 82-5). Together, these complexes generate a small amount of thrombin that feeds back to amplify its own generation by activating the nonenzyme cofactors and platelets. The phosphatidylserine expressed on the surface of activated platelets provides an anionic surface on which the complexes assemble. The three enzyme complexes involved in thrombin generation are extrinsic tenase, intrinsic tenase, and prothrombinase. Although extrinsic tenase initiates the system under most circumstances, the contact system also plays a role in some situations.
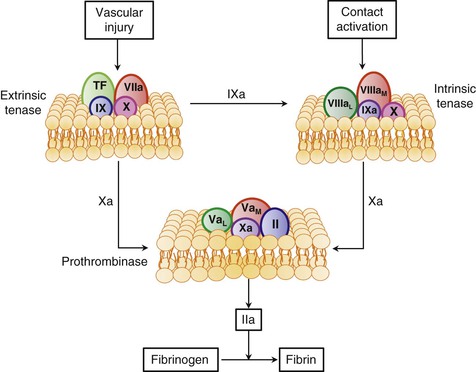
Extrinsic Tenase
This complex forms on exposure of tissue factor–expressing cells to blood. Tissue factor exposure occurs after atherosclerotic plaque rupture because the core of the plaque is rich in cells that express tissue factor. Denuding injury to the vessel wall also exposes the tissue factor constitutively expressed by subendothelial fibroblasts and smooth muscle cells. In addition to cells in the vessel wall, circulating monocytes and monocyte-derived microparticles (small membrane fragments) also provide a source of tissue factor.20 When tissue factor–bearing monocytes or microparticles bind to platelets or other leukocytes and their plasma membranes fuse, transfer of tissue factor takes place. By binding to the adhesion molecules expressed on activated endothelial cells or to P-selectin on activated platelets, these tissue factor–bearing cells or microparticles can initiate or augment coagulation.21 This phenomenon probably explains how venous thrombi develop in the absence of obvious vessel wall injury.2
The integral membrane protein tissue factor serves as a receptor for factor VIIa. Blood contains trace amounts of factor VIIa, which has negligible activity in the absence of tissue factor.22 With exposure of tissue factor on anionic cell surfaces, factor VIIa binds in a calcium-dependent fashion to form the extrinsic tenase complex, which is a potent activator of factors IX and X. Once activated, factors IXa and Xa serve as the enzyme components of intrinsic tenase and prothrombinase, respectively.
Intrinsic Tenase
Factor IXa binds to factor VIIIa on anionic cell surfaces to form the intrinsic tenase complex. Factor VIII circulates in blood in a complex with vWF. Thrombin cleaves factor VIII and releases it from vWF, thereby converting it to its activated form. Activated platelets express binding sites for factor VIIIa. Once bound, factor VIIIa binds factor IXa in a calcium-dependent fashion to form the intrinsic tenase complex, which then activates factor X. The change in catalytic efficiency of factor IXa–mediated activation of factor X that occurs with deletion of individual components of the intrinsic tenase complex highlights their importance. Absence of the membrane or factor VIIIa almost completely abolishes enzymatic activity, and the catalytic efficiency of the complete complex is 109-fold greater than that of factor IXa alone. Because intrinsic tenase activates factor X at a rate 50- to 100-fold faster than extrinsic tenase does, intrinsic tenase plays a critical role in the amplification of factor Xa and thrombin generation. The bleeding that occurs in patients with hemophilia—congenital deficiency of factor VIII or factor IX—highlights the importance of intrinsic tenase in hemostasis.
Prothrombinase
Factor Xa binds to factor Va, its activated cofactor, on anionic phospholipid membrane surfaces to form the prothrombinase complex. Activated platelets release factor V from their alpha granules, and this platelet-derived factor V may play a more important role in hemostasis than its plasma counterpart does. Although plasma factor V requires thrombin activation to exert its cofactor activity, the partially activated factor V released from platelets already exhibits substantial cofactor activity. Activated platelets express specific factor Va binding sites on their surface, and bound factor Va serves as a receptor for factor Xa. The catalytic efficiency of activation of prothrombin by factor Xa increases by 109-fold when factor Xa is incorporated into the prothrombinase complex. Prothrombin binds to the prothrombinase complex, where it undergoes conversion to thrombin in a reaction that releases prothrombin fragment 1.2 (F1.2). Plasma levels of F1.2 therefore provide a marker of prothrombin activation.
Fibrin Formation
The final effector in coagulation is thrombin. Thrombin converts soluble fibrinogen to insoluble fibrin. Fibrinogen is a dimeric molecule, each half of which is composed of three polypeptide chains—the Aα, Bβ, and γ chains. Numerous disulfide bonds covalently link the chains together and join the two halves of the fibrinogen molecule (Fig. 82-6). Electron micrographic studies of fibrinogen reveal a trinodular structure with a central E domain flanked by two D domains. Crystal structures show symmetry of design with the central E domain, which contains the amino-terminals of the fibrinogen chains joined to the lateral D domains by coiled-coil regions.
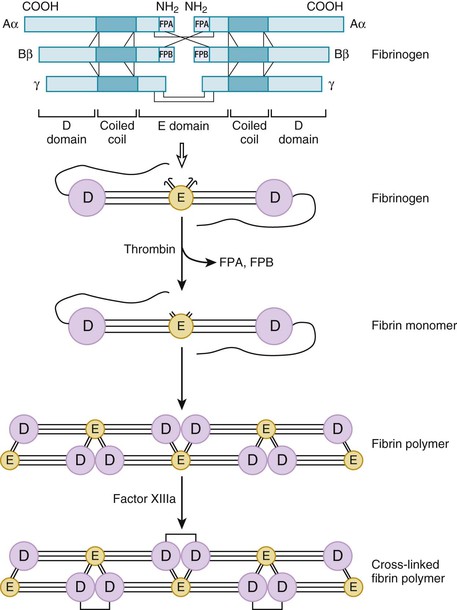
Fibrinogen, the most abundant plasma protein involved in coagulation, circulates in an inactive form. Thrombin binds to the amino-terminals of the Aα and Bβ chains of fibrinogen, where it cleaves specific peptide bonds to release fibrinopeptide A and fibrinopeptide B and generates fibrin monomer (Fig. 82-6). Because they are products of the action of thrombin on fibrinogen, plasma levels of these fibrinopeptides provide an index of thrombin activity. Release of fibrinopeptide creates new amino-terminals that extend as knobs from the E domain of one fibrin monomer and insert into preformed holes in the D domains of other fibrin monomers. This creates long strands known as protofibrils that consist of fibrin monomers noncovalently linked together in a half-staggered overlapping fashion.
Noncovalently linked fibrin protofibrils are unstable. By covalently cross-linking the α and γ chains of adjacent fibrin monomers, factor XIIIa stabilizes the fibrin in a calcium-dependent fashion and renders it relatively resistant to degradation. Factor XIII circulates in blood as a heterodimer consisting of two A and two B subunits. The active site and calcium binding site of factor XIII are localized to the A subunit. Platelets contain large amounts of factor XIII in their cytoplasm, but platelet-derived factor XIII consists only of A subunits. Both plasma factor XIII and platelet factor XIII are activated by thrombin.
Contact Pathway
Current thinking is that exposure of tissue factor represents the sole pathway for activation of coagulation and that the contact system—which includes factor XII, prekallikrein, and high-molecular-weight kininogen—is unimportant for hemostasis because patients deficient in these factors do not have bleeding problems. The physiologic role of factor XI is more difficult to assess because the plasma level of factor XI in patients with congenital deficiency of factor XI, so-called hemophilia C, does not predict the propensity for bleeding. Although the capacity of thrombin to feed back and activate platelet-bound factor XI may explain this phenomenon, platelet-derived factor XI may be more important than circulating factor XI for hemostasis.
We cannot ignore the contact pathway, however, because coronary catheters and other blood-contacting medical devices, such as stents or mechanical valves, probably trigger clotting through this mechanism.23 Factor XII binds to the surface of catheters or devices, where it undergoes a conformational change that results in its activation. Factor XIIa converts prekallikrein to kallikrein in a reaction accelerated by high-molecular-weight kininogen, and factor XIIa and kallikrein then feed back to activate additional factor XII. Factor XIIa propagates coagulation by activating factor XI (Fig. 82-7).
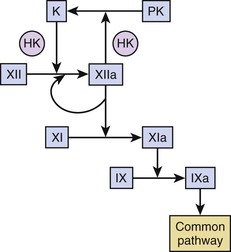
In addition to its role in device-related thrombosis, the contact pathway may also contribute to the stabilization of arterial and venous thrombi. Thus mice deficient in factor XII or factor XI form small unstable thrombi at sites of arterial or venous damage, thus suggesting that these factors contribute to thrombus stabilization.24 Potential physiologic activators of the contact pathway include polyphosphates released from activated platelets, DNA or RNA released from damaged or apoptotic cells in atherosclerotic plaque, and the DNA and histone network of NETs extruded from activated neutrophils, which not only promotes platelet adhesion and activation but also triggers activation of factor XII. Support for the role of these activators in thrombosis comes from observations in mice that phosphatases and DNA- or RNA-degrading enzymes attenuate thrombosis at sites of injury. The role of these activators in thrombosis in humans is unclear. Although patients with unstable angina have increased plasma levels of factor XIa,25 it is unknown whether this reflects activation by factor XIIa or by thrombin.
Regardless of the extent to which the contact pathway contributes to thrombin generation, the final product of coagulation is fibrin. Hemostasis depends on a dynamic balance between the formation of fibrin and its degradation; the fibrinolytic system mediates fibrin breakdown.
Fibrinolytic System
Fibrinolysis is initiated when plasminogen activators convert plasminogen to plasmin, which then degrades fibrin into soluble fragments (Fig. 82-8). Blood contains two immunologically and functionally distinct plasminogen activators, t-PA and u-PA. t-PA mediates intravascular fibrin degradation, whereas u-PA binds to a specific u-PA receptor (u-PAR) on the surface of cells, where it activates cell-bound plasminogen.10 Consequently, pericellular proteolysis during cell migration and tissue remodeling and repair are the major functions of u-PA.
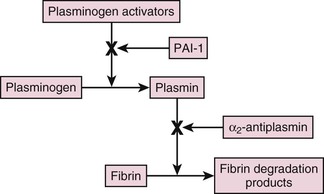
Regulation of fibrinolysis occurs at two levels. PAI-1 and, to a lesser extent, PAI-2 inhibit the plasminogen activators, whereas alpha2-antiplasmin inhibits plasmin. Endothelial cells synthesize PAI-1, which inhibits both t-PA and u-PA, whereas monocytes and the placenta synthesize PAI-2, which specifically inhibits u-PA.10 Thrombin-activated fibrinolysis inhibitor (TAFI) also modulates fibrinolysis and provides a link between fibrinolysis and coagulation.26 Thrombosis can occur if activation of the fibrinolytic system is impaired, whereas excessive activation leads to bleeding. Therefore a review of the mechanisms of action of t-PA, u-PA, and TAFI is worthwhile.
Mechanism of Action of Tissue Plasminogen Activator
t-PA, a serine protease, contains five discrete domains: a fibronectin-like finger domain, an epidermal growth factor domain, two kringle domains, and a protease domain. Synthesized as a single-chain polypeptide, plasmin converts single-chain t-PA into a two-chain form. Both forms of t-PA convert plasminogen to plasmin. Native Glu-plasminogen is a single-chain polypeptide with a Glu residue at its amino-terminal. Plasmin cleavage at the amino-terminal generates Lys-plasminogen, a truncated form with a Lys residue at its new amino-terminal. t-PA cleaves a single peptide bond to convert single-chain Glu- or Lys-plasminogen into two-chain plasmin, which is composed of a heavy chain containing five kringle domains and a light chain containing the catalytic domain. Because its open conformation exposes the t-PA cleavage site, Lys-plasminogen is a better substrate for t-PA than Glu-plasminogen is, which assumes a circular conformation that renders this bond less accessible.
t-PA has little enzymatic activity in the absence of fibrin, but its activity increases by at least three orders of magnitude when fibrin is present.10 This increase in activity reflects the capacity of fibrin to serve as a template that binds t-PA and plasminogen and promotes their interaction. t-PA binds to fibrin via its finger and second kringle domains, whereas plasminogen binds fibrin via its kringle domains. Kringle domains are looplike structures that bind Lys residues on fibrin. As fibrin undergoes degradation, more Lys residues are exposed, which provides additional binding sites for t-PA and plasminogen. Consequently, degraded fibrin stimulates activation of plasminogen by t-PA more than intact fibrin does.
Alpha2-antiplasmin rapidly inhibits circulating plasmin by docking to its first kringle domain and then inhibiting the active site.10 Because plasmin binds to fibrin via its kringle domains, plasmin generated on the fibrin surface resists inhibition by alpha2-antiplasmin. This phenomenon endows fibrin-bound plasmin with the capacity to degrade fibrin. Factor XIIIa cross-links small amounts of alpha2-antiplasmin onto fibrin, which prevents premature fibrinolysis.
Like fibrin, annexin II on endothelial cells binds t-PA and plasminogen and promotes their interaction. Cell surface gangliosides and alpha-enolase may also bind plasminogen and promote its activation by altering its conformation into the more readily activated open form. Plasminogen binds to endothelial cells via its kringle domains. Lipoprotein(a), which also possesses kringle domains, impairs cell-based fibrinolysis by competing with plasminogen for cell surface binding (see also Chapter 45). This phenomenon may explain the association between elevated levels of lipoprotein(a) and atherosclerosis (see also Chapters 42 and 45).27
Mechanism of Action of Urokinase Plasminogen Activator
Synthesized as a single-chain polypeptide, single-chain u-PA (scu-PA) has minimal enzymatic activity. Plasmin readily converts scu-PA into a two-chain form that is enzymatically active and capable of binding u-PAR on cell surfaces. Further cleavage at the amino-terminals of two-chain u-PA yields a truncated, lower-molecular-weight form that lacks the u-PAR binding domain.10
Two-chain forms of u-PA readily convert plasminogen to plasmin in the absence or presence of fibrin. In contrast, scu-PA does not activate plasminogen in the absence of fibrin, but it can activate fibrin-bound plasminogen because plasminogen adopts a more open and readily activatable conformation when immobilized on fibrin. Like the higher-molecular-weight form of two-chain u-PA, scu-PA binds to cell surface u-PAR, where plasmin can activate it. Many tumor cells elaborate u-PA and express u-PAR on their surface. Plasmin generated on these cells endows them with the capacity for metastasis.28
Mechanism of Action of Thrombin-Activated Fibrinolysis Inhibitor
TAFI, a procarboxypeptidase B–like molecule synthesized in the liver, circulates in blood in a latent form, where thrombin bound to thrombomodulin can activate it. Unless bound to thrombomodulin, thrombin activates TAFI inefficiently.26 Activated TAFI (TAFIa) attenuates fibrinolysis by cleaving Lys residues from the carboxy-terminals of chains of degrading fibrin, thereby removing binding sites for plasminogen, plasmin, and t-PA. TAFI links fibrinolysis to coagulation in that the thrombin-thrombomodulin complex not only activates TAFI, which attenuates fibrinolysis, but also activates protein C, which mutes thrombin generation.
TAFIa has a short half-life in plasma because the enzyme is unstable.26 Genetic polymorphisms can result in the synthesis of more stable forms of TAFIa. Persistent attenuation of fibrinolysis by these variant forms of TAFIa may render patients susceptible to thrombosis.
Thrombosis
A physiologic host defense mechanism, hemostasis focuses on arrest of bleeding by forming hemostatic plugs composed of platelets and fibrin at sites of vessel injury. In contrast, thrombosis reflects a pathologic process associated with intravascular thrombi that fill the lumens of arteries or veins.
Arterial Thrombosis (See Chapter 41)
Most arterial thrombi occur on top of disrupted atherosclerotic plaque. Coronary plaque with a thin fibrous cap and a lipid-rich core is most prone to disruption.1 Rupture of the fibrous cap exposes thrombogenic material in the lipid-rich core to blood and triggers platelet activation and thrombin generation. The extent of plaque disruption and the content of thrombogenic material in the plaque determine the consequences of the event, but host factors also contribute. Breakdown of the regulatory mechanisms that limit platelet activation and inhibit coagulation can augment thrombosis at sites of plaque disruption.
Decreased production of nitric oxide and prostacyclin by diseased endothelial cells can trigger vasoconstriction and platelet activation.29 Proinflammatory cytokines lower expression of thrombomodulin by endothelial cells, which promotes thrombin generation, and stimulate expression of PAI-1, which inhibits fibrinolysis.30
Products of blood coagulation contribute to atherogenesis, as well as to its complications. Microscopic erosions in the vessel wall trigger the formation of tiny platelet-rich thrombi. Activated platelets release PDGF and TGF-β, which promote a fibrotic response.31 Thrombin generated at the site of injury not only activates platelets and converts fibrinogen to fibrin but also activates PAR-1 on smooth muscle cells and induces their proliferation, migration, and elaboration of extracellular matrix. Incorporation of microthrombi into plaque promotes their growth, and decreased endothelial cell production of heparan sulfate—which normally limits smooth muscle proliferation—contributes to plaque expansion. The multiple links between atherosclerosis and thrombosis have prompted the term atherothrombosis.
Venous Thrombosis (Also see Chapter 73)
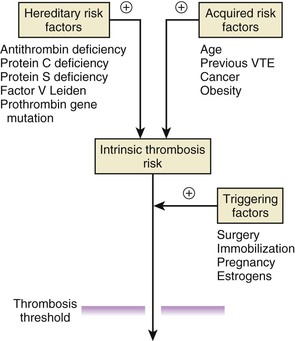
Causes of venous thrombosis include those associated with hypercoagulability, which can be genetic or acquired, and the mainly acquired risk factors, such as advanced age, obesity, or cancer, which are associated with immobility (Table 82-1). Inherited hypercoagulable states and these acquired risk factors combine to establish the intrinsic risk for thrombosis in each individual. Superimposed triggering factors, such as surgery, pregnancy, or hormonal therapy, modify this risk, and thrombosis occurs when the combination of genetic, acquired, and triggering forces exceeds a critical threshold (Fig. 82-9).
TABLE 82-1
Classification of Hypercoagulable States
| HEREDITARY | MIXED | ACQUIRED |
| Loss of Function | ||
| Antithrombin deficiency | Hyperhomocysteinemia | Advanced age |
| Protein C deficiency | Previous venous thromboembolism | |
| Protein S deficiency | Surgery | |
| Gain of Function | Immobilization | |
| Factor V Leiden | Obesity | |
| Prothrombin gene mutation | Cancer | |
| Elevated factor VIII, IX, or XI levels | Pregnancy, puerperium Drug-induced: l– asparaginase, hormonal therapy | |
Some acquired or triggering factors entail higher risk than do others. For example, major orthopedic surgery, neurosurgery, multiple trauma, and metastatic cancer (particularly adenocarcinoma) are associated with the highest risk, whereas prolonged bed rest, antiphospholipid antibodies, and the puerperium are associated with intermediate risk; pregnancy, obesity, long-distance travel, and the use of oral contraceptives or hormonal replacement therapy are mild risk factors. Up to half of patients with VTE before the age of 45 years have inherited hypercoagulable disorders—so-called thrombophilia—particularly those whose event occurred in the absence of risk factors or with minimal provocation, such as after minor trauma or a long-haul flight or with estrogen use. The following sections describe the inherited and acquired hypercoagulable states.
Inherited Hypercoagulable States
Inherited hypercoagulable states fall into two categories. Some are associated with gain-of-function mutations in procoagulant pathways, such as factor V Leiden, the prothrombin gene mutation, and increased levels of procoagulant proteins; others are associated with loss-of-function mutations of endogenous anticoagulant proteins, such as deficiencies of antithrombin, protein C, and protein S. Although all these inherited hypercoagulable disorders increase the risk for VTE, only increased levels of procoagulant proteins are clearly associated with increased risk for arterial thrombosis.
Factor V Leiden
The factor V Leiden mutation, present in about 5% of white individuals, is the most common inherited thrombophilia. Because of a founder effect, the mutation is less common in Hispanics and blacks and rare in Asians. Caused by a point mutation in the factor V gene, the defect results in the synthesis of a factor V molecule with a Gln residue in place of an Arg residue at position 506—one of three sites where activated protein C cleaves factor Va to inactivate it. Consequently, activated factor V Leiden resists rapid proteolysis and persists 10-fold longer in the presence of activated protein C than its wild-type counterpart does. Inherited in an autosomal dominant fashion, individuals heterozygous for the factor V Leiden mutation have a fivefold increased risk for VTE; those homozygous for the mutation have higher risk. However, the absolute risk for venous thrombosis is low with factor V Leiden, and with a yearly risk of 0.1% to 0.3%, subjects with this disorder have a lifetime risk for thrombosis of only 5% to 10%.
An activated protein C resistance assay establishes the diagnosis of factor V Leiden in most cases. This assay involves calculation of the ratio of the activated partial thromboplastin time (APTT) measured after the addition of activated protein C divided by that determined before its addition. Use of factor V–deficient plasma increases the specificity of the test. If the result of the clotting assay is equivocal, genetic testing using a polymerase chain reaction (PCR)-based assay confirms the diagnosis.
Prothrombin Gene Mutation
The second most common thrombophilic disorder, the prothrombin gene mutation, reflects a G-to-A nucleotide transition at position 20210 in the 3′-untranslated region of the prothrombin gene. This mutation causes elevated levels of prothrombin, which enhances thrombin generation and may limit inactivation of factor Va by activated protein C. The exact mechanism by which the G20210A mutation increases prothrombin levels remains controversial. Enhanced protein synthesis may result from more efficient 3′-end formation, increased messenger RNA stability, enhanced translation efficiency, or some combination of these mechanisms.
The prevalence of the prothrombin gene mutation is about 3% in white persons and lower in Asians and blacks. The mutation increases the risk for venous thrombosis to a similar extent as factor V Leiden does. Laboratory diagnosis depends on genetic screening after PCR amplification of the 3′-untranslated region of the prothrombin gene. Although heterozygotes have 30% higher levels of prothrombin than noncarriers do, the wide range of prothrombin levels in healthy individuals precludes the use of this phenotype for carrier identification.
Elevated Levels of Procoagulant Proteins
Elevated levels of factor VIII and other coagulation factors, including fibrinogen and factors IX and XI, appear to be independent risk factors for venous thrombosis. Increased levels of factor VIII have also been associated with an up to threefold increase in the risk for myocardial infarction.32 Although the molecular bases for the high levels of these coagulation factors have yet to be identified, genetic mechanisms probably contribute because these quantitative abnormalities have high heritability.
Antithrombin Deficiency
Synthesized in the liver, antithrombin regulates coagulation by forming a 1:1 covalent complex with thrombin, factor Xa, or other activated clotting factors. Heparan sulfate or heparin accelerates the rate of antithrombin interaction with its target proteases. Inherited antithrombin deficiency is rare; it occurs in approximately 1 in 2000 people and can be due to decreased synthesis of a normal protein or synthesis of a dysfunctional protein. A parallel reduction in the levels of antithrombin antigen and activity identifies deficiencies caused by decreased synthesis, whereas decreased antithrombin activity in the presence of normal antigen levels identifies dysfunctional forms of antithrombin. A measurement of antithrombin activity with or without added heparin identifies variants with impaired heparin-binding capacity.
Acquired antithrombin deficiency results from decreased synthesis, increased consumption, or enhanced clearance. Decreased synthesis can occur in patients with severe hepatic disease, particularly cirrhosis, or in those given l-asparaginase. Increased activation of coagulation can result in antithrombin consumption in disorders such as extensive thrombosis, disseminated intravascular coagulation, severe sepsis, disseminated malignancy, or prolonged extracorporeal circulation. Heparin treatment can also reduce antithrombin levels up to 20% by enhancing its clearance. Severe antithrombin deficiency can develop in some patients with nephrotic syndrome because of loss of protein in urine.
Protein C Deficiency
Thrombin initiates the protein C pathway when it binds thrombomodulin on the endothelial cell surface (see Fig. 82-2). Thrombin bound to thrombomodulin activates protein C approximately 1000-fold more efficiently than free thrombin does.9 The EPCR augments this process 20-fold by binding protein C and presenting it to the thrombin-thrombomodulin complex for activation.9 Activated protein C then dissociates from the activation complex and decreases thrombin generation by inactivating factors Va and VIIIa on the activated platelet surface. For efficient inactivation of these factors, activated protein C must bind to protein S, its cofactor.
Protein C deficiency can be caused by both inherited and acquired forms. Approximately 1 in 200 adults has heterozygous protein C deficiency inherited in an autosomal dominant fashion, but most have no history of thrombosis. The variable phenotypic expression of hereditary protein C deficiency suggests the existence of other, yet unrecognized modifying factors. In contrast to antithrombin deficiency, in which the homozygous state is associated with embryonic lethality, homozygous or doubly heterozygous protein C deficiency can occur. Newborns with these disorders often have purpura fulminans characterized by widespread thrombosis.
Inherited protein C deficiency can result from decreased synthesis of normal protein or from synthesis of dysfunctional forms of protein C. Identification of the type of deficiency requires simultaneous measurement of protein C antigen and activity; reduced synthesis of a normal protein results in a parallel reduction in protein C antigen and activity, whereas synthesis of a dysfunctional protein results in normal antigen with reduced activity.
Acquired protein C deficiency can be due to decreased synthesis or increased consumption. Decreased synthesis can occur in patients with liver disease or in those given warfarin. Protein C consumption can occur with severe sepsis, with disseminated intravascular coagulation, and after surgery. Although antithrombin levels can be low in patients with nephrotic syndrome, protein C levels are normal or elevated in such patients.
Protein S Deficiency
Protein S serves as a cofactor for activated protein C (see Fig. 82-3). In addition, protein S may directly inhibit prothrombin activation because of its capacity to bind factor Va and/or factor Xa, components of the prothrombinase complex. The importance of the direct anticoagulant activity of protein S is uncertain.
In the circulation, approximately 60% of total protein S is bound to C4b-binding protein, a complement component; only the remaining free 40% is functionally active. Diagnosis of protein S deficiency requires measurement of both the free and bound forms of protein S. Inherited protein S deficiency can result from reduced synthesis of the protein or synthesis of a dysfunctional protein. Acquired protein S deficiency can be due to decreased synthesis, increased consumption, loss, or shift of free protein S to the bound form. Decreased synthesis can occur in patients with severe liver disease or in those given warfarin or l-asparaginase. Increased consumption of protein S occurs in patients with acute thrombosis or disseminated intravascular coagulation. Patients with nephrotic syndrome can excrete free protein S in their urine, which causes decreased protein S activity. Total protein S levels in these patients are often normal because the levels of C4b-binding protein increase, thus shifting more protein S to the bound form. C4b-binding protein levels also increase in pregnancy and with the use of oral contraceptives. This shifts more protein S to the bound form and lowers the levels of free protein S and protein S activity. The consequences of this phenomenon are uncertain.
Other Hereditary Disorders
A polymorphism in the gene that encodes the EPCR has been linked to venous thrombosis. Associated with EPCR shedding and high levels of soluble EPCR, this polymorphism reduces endothelial EPCR, and soluble EPCR competes with its endothelial cell counterpart for protein C binding.33
A polymorphism in factor XIII that results in more rapid activation by thrombin has been associated with a small reduction in the risk for VTE, myocardial infarction, and ischemic stroke in some case-control studies but not in others.34 The frequency of this polymorphism varies among different ethnic populations, and certain environmental factors, such as obesity and estrogen therapy, may augment its protective effect. More work is needed to determine the extent to which this polymorphism modulates the risk for thrombosis.
Acquired Hypercoagulable States
Such states include surgery and immobilization, advanced age, obesity, cancer, pregnancy and estrogen therapy (oral contraceptive or hormone replacement therapy), a previous history of VTE, antiphospholipid antibody syndrome, and hyperhomocysteinemia (Table 82-1). These conditions can occur in isolation or in conjunction with hereditary hypercoagulable states.
Surgery and Immobilization
Surgery can directly damage veins, and immobilization after surgery leads to stasis in the deep veins of the leg. The risk for VTE in surgical patients depends on the patient’s age, the type of surgery, and the presence of active cancer. Patients older than 65 years are at greater risk, and high-risk types of surgery include major orthopedic procedures, neurosurgery, and extensive abdominal or pelvic surgery, especially for cancer. Because the risk for VTE increases up to 20-fold in these patients, they require vigorous thromboprophylaxis until they are fully mobile.
Hospitalization and nursing home confinement account for approximately 60% of cases of VTE, again reflecting the impact of immobilization. Hospitalization for medical illness accounts for a similar proportion of cases as hospitalization for surgery, thus highlighting the need for thromboprophylaxis in medical patients as well as in surgical patients.
Advanced Age
Predominantly a disease of older age, VTE in those younger than 50 years has an incidence of 1 per 10,000 and increases approximately 10-fold per decade thereafter. Men have an overall age-adjusted incidence rate approximately 1.2-fold higher than women do. Although incidence rates are higher in women during the reproductive years, after 45 years of age, men have higher incidence rates. Many potential mechanisms may increase the incidence of VTE with advanced age, including decreased mobility, intercurrent diseases, and vascular endothelium that is less resistant to thrombosis. Levels of procoagulant proteins also increase with age.
Obesity
The risk for VTE increases approximately 1.2-fold for every 10-kg/m2 increase in body mass index, but the basis for the association between obesity and VTE is unclear. Obesity leads to immobility; in addition, adipose tissue, particularly visceral fact, expresses proinflammatory cytokines and adipokines, which may promote coagulation by increasing levels of procoagulant proteins or impair fibrinolysis by elevating levels of PAI-1. With the growing epidemic of obesity, the incidence of VTE may increase.
Cancer
Approximately 20% of patients with VTE have cancer.35 Cancer patients in whom VTE develops have reduced survival in comparison to those without VTE. Patients with brain tumors and advanced ovarian or prostate cancer have particularly high rates of VTE. Treatment with chemotherapy, hormonal therapy, and biologic agents (such as erythropoietin and antiangiogenic drugs) further increases the risk, as do central venous catheters and surgery for cancer.
The pathogenesis of thrombosis in cancer patients is multifactorial in origin and represents a complex interplay between the tumor, patient characteristics, and the hemostatic system. Many types of tumor cells express tissue factor or other procoagulants that can initiate coagulation. In addition to its role in coagulation, tissue factor also acts as a signaling molecule that promotes tumor proliferation and spread.36
Patient characteristics that contribute to VTE include immobility and venous stasis secondary to extrinsic compression of major veins by tumor. Surgical procedures, central venous catheters, and chemotherapy can injure vessel walls. In addition, tamoxifen and selective estrogen receptor modulators (SERMs) induce an acquired hypercoagulable state by reducing levels of natural anticoagulant proteins.
A proportion of patients with unprovoked VTE have occult cancer. This observation has prompted some experts to recommend extensive screening for cancer in such patients, but the potential harm—including procedure-related morbidity, the psychological impact of false-positive test results, and the cost of screening—offsets any benefits of this approach. Small studies comparing extensive cancer screening with no screening in patients with unprovoked VTE have not demonstrated a reduction in cancer-related mortality with screening. Therefore unless symptoms suggestive of underlying cancer are present, only age-appropriate screening for breast, cervical, colon, and possibly prostate cancer is indicated because screening for these cancers may reduce mortality.
Pregnancy
Pregnant women have a fivefold to sixfold higher risk for VTE than do age-matched nonpregnant women. VTE occurs in approximately 1 in 1000 pregnancies, and in approximately 1 in 1000 women VTE develops in the postpartum period. VTE is the leading cause of maternal morbidity and mortality. Patient-related factors influence the risk for VTE in pregnancy and the puerperium, including age older than 35 years, body mass index higher than 29, cesarean delivery, thrombophilia, or a personal or family history of VTE. Ovarian hyperstimulation and multiparity are other risk factors.
More than 90% of deep vein thrombi in pregnancy occur in the left leg, probably because the enlarged uterus compresses the left iliac vein by exerting pressure on the overlying right iliac and ovarian arteries. Hypercoagulability occurs in pregnancy because of the combination of venous stasis and changes in blood. Uterine enlargement reduces venous blood flow from the lower extremities. This is not the only mechanism responsible for venous stasis, however, because blood flow from the lower extremities begins to decrease by the end of the first trimester, probably as a result of hormonally induced venous dilation. Systemic factors also contribute to hypercoagulability. Thus levels of procoagulant proteins, such as factor VIII, fibrinogen, and vWF, increase in the third trimester of pregnancy. Coincidentally, suppression of the natural anticoagulant pathways takes place. The net effect of these changes is enhanced thrombin generation, as evidenced by elevated levels of F1.2 and thrombin-antithrombin complexes.
About half the episodes of VTE in pregnancy occur in women with thrombophilia. The risk for VTE in women with thrombophilic defects depends on the type of abnormality and the presence of other risk factors. Risk appears to be highest in women with antithrombin, protein C, or protein S deficiency and lower in those with factor V Leiden or the prothrombin gene mutation. In general, the daily risk for VTE in these women is higher in the postpartum period than it is during pregnancy. The risk during pregnancy is similar in all three trimesters. Therefore women needing thromboprophylaxis require treatment throughout pregnancy and for at least 6 weeks postpartum.
Sex Hormone Therapy (Also see Chapter 77)
Oral contraceptives, estrogen replacement therapy, and SERMs are all associated with an increased risk for venous thrombosis. The relatively high risk for VTE associated with first-generation oral contraceptives prompted the development of low-dose formulations. Currently available low-estrogen combination oral contraceptives contain 20 to 50 µg of ethinylestradiol and one of several different progestins. Even these low-dose combination contraceptives are associated with a threefold to fourfold increased risk for VTE in comparison to nonusers. In absolute terms this translates to an incidence of 3 to 4 per 10,000 as compared with 5 to 10 per 100,000 in nonusers of reproductive age.
Even though smoking increases the risk for myocardial infarction and stroke in women taking oral contraceptives, it is unclear whether smoking affects the risk for VTE. Obesity, however, affects the risk for both arterial and venous thrombosis. The risk for VTE is highest during the first year of oral contraceptive use and persists only for the duration of use.
Case-control studies suggest a 20- to 30-fold higher risk for VTE in women with inherited thrombophilia who use oral contraceptives than in nonusers with thrombophilia or users without these defects. Despite the increased risk, routine screening for thrombophilia in young women considering oral contraceptive use is not necessary. Based on the incidence and case fatality rate of thrombotic events, estimates suggest that screening 400,000 women would detect 20,000 factor V Leiden carriers and that prevention of a single death would necessitate withholding oral contraceptives in all these women. Even larger numbers of women with less prevalent thrombophilic defects would require screening. Based on these considerations, routine screening is not recommended.
Hormonal replacement therapy with conjugated equine estrogen, with or without a progestin, is associated with a small increase in the risk for myocardial infarction, ischemic stroke, and VTE. SERMs, such as tamoxifen, are estrogen-like compounds that serve as an estrogen antagonist in the breast but as estrogen agonists in other tissues, such as bone and the uterus. Like estrogens, tamoxifen increases the risk for VTE by threefold to fourfold. The risk is higher in postmenopausal women, particularly those receiving systemic combination chemotherapy. Because of this risk, aromatase inhibitors, which antagonize estrogens by blocking their synthesis from androgens, are replacing tamoxifen for the treatment of estrogen receptor–positive breast cancer. These newer agents are associated with a lower risk for VTE than tamoxifen is. Raloxifene, a SERM used to prevent osteoporosis, increases the risk for VTE threefold when compared with placebo, which contraindicates the use of raloxifene for prevention of osteoporosis in women with a history of VTE.
History of Previous Venous Thromboembolism
A history of previous VTE places patients at risk for recurrence. When anticoagulation treatment stops, patients with unprovoked VTE have a risk for recurrence of approximately 10% at 1 year and 30% at 5 years. This risk appears to be independent of whether an underlying thrombophilic defect is present, such as factor V Leiden or the prothrombin gene mutation.
The risk for recurrent VTE is lower in patients whose incident event occurred in association with a transient risk factor, such as major surgery or prolonged immobilization. These patients have a risk for recurrence of approximately 4% at 1 year and 10% at 5 years. Patients whose VTE occurred on the background of minor risk factors, such as oral contraceptive use or a long-haul flight, probably have an intermediate risk for recurrence. Patients at highest risk for recurrence are those with inherited deficiencies of antithrombin, protein C, or protein S; those with antiphospholipid antibody syndrome; patients with advanced malignancy; or those homozygous for factor V Leiden or the prothrombin gene mutation. Their risk for recurrence is likely to be 15% at 1 year and up to 50% at 5 years.
Antiphospholipid Antibody Syndrome
A heterogeneous group of autoantibodies directed against proteins that bind phospholipid, antiphospholipid antibodies can be categorized into those that prolong phospholipid-dependent coagulation assays, so-called lupus anticoagulants (LAs), or anticardiolipin (ACL) antibodies, which target cardiolipin. A subset of ACL antibodies recognizes other phospholipid-bound proteins, particularly beta2-glycoprotein I.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


