Chapter 5 Hemostasis and Thrombosis
Hemostasis
Components of Hemostasis
Vessel Response
In normal vessels, endothelial cells cover the luminal surface, forming a monolayer with tight cell-cell interaction.1 Once regarded as a passive barrier between the blood and the underlying thrombogenic subendothelium, the endothelium is now recognized as a biologically active organ that participates in and modulates various physiologic processes, including hemostasis and thrombosis.
In their quiescent state, endothelial cells are actively antithrombotic (Box 5-1). They synthesize and secrete several modulators that lead to vasodilation, decreased platelet aggregation, decreased levels of thrombin, factors Va and VIIIa, and factors IXa and Xa by which an antithrombotic state is promoted. Specifically, prostacyclin and nitric oxide are potent vasodilators and inhibitors of platelet aggregation. Heparan sulfates accelerate the activity of antithrombin (AT), thereby inactivating thrombin. Thrombomodulin (TM) also inactivates thrombin2 by forming the thrombomodulin-thrombin complex, a potent activator of protein C,3 which with the help of cofactor protein S inactivates factors Va and VIIIa, leading to decreased thrombin and factor Xa levels. Tissue factor pathway inhibitor (TFPI), which is bound to the endothelial surface, strongly inhibits the external coagulation pathway after heparin administration.4,5 Tissue-type plasminogen activator (t-PA) and urokinase are synthesized, which bind fibrinogen and fibrin, increase plasmin, and promote fibrinolysis.
Box 5-1
Endothelial Cell as Modulator of Hemostasis
Function Effect
Thrombogenic
• Profound loss of NO and PGI2 after injury
• Loss of vasodilating stimulus
• Von Willebrand factor synthesis: ↑Platelet adhesion
• Factor V synthesis: ↑Thrombin
• Expression of tissue factor: ↑Thrombin
• Binding of factors VIIa and IXa: ↑Thrombin
• Surface membrane site for prothrombinase complex: ↑Thrombin
Antithrombotic
• PGI2 synthesis and granule release: ↓Platelet aggregation
• Thrombomodulin synthesis: ↓Factors Va and VIIIa
• Protein S synthesis: ↓Factors Va and VIIIa
• Heparan sulfate synthesis: ↓Thrombin
• t-PA and urokinase synthesis: ↓Plasmin
NO, Nitric oxide; PGI2, prostaglandin I2; t-PA, tissue plasminogen activator.
The endothelium also possesses substantial procoagulant activity and acts as a scaffold for hemostasis when stimulated after vessel injury (see Box 5-1). Tissue factor (thromboplastin, factor III) is a lipoprotein that is constitutively expressed by most cells; however, endothelial cells only express tissue factor when stimulated by agonists such as thrombin or endotoxin. Vessel injury causes endothelial denudation and activation, which result in exposure of tissue factor to low circulating levels of activated factor VII in blood to form complexes that catalyze the conversion of factor IX to IXa and factor X to Xa, leading to thrombin formation.
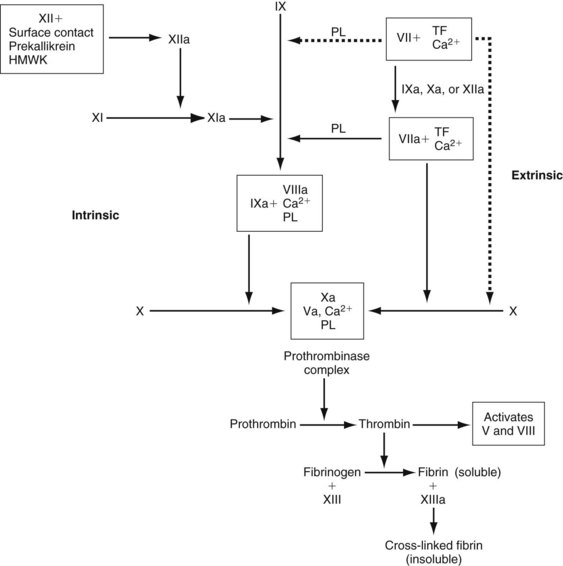
Endothelial cells also synthesize and secrete von Willebrand factor (vWF), which is necessary for platelet adhesion to the vessel wall. This factor has binding sites for collagen, platelet glycoproteins (GPs) Ib and IIb/IIIa, and factor VIII. Factor VIII and vWF circulate together as a complex. Endothelial cells, in addition to the liver, synthesize factor V. Factors V and VIII are cleaved by thrombin into their activated states (Va and VIIIa) and then become integral components of membrane-bound complexes that accelerate the formation of thrombin and factor Xa (Figure 5-1). Endothelial cells also synthesize plasminogen activator inhibitor (PAI-1), which rapidly inactivates circulating t-PA.
Platelet Activation
The platelet membrane is composed of a phospholipid bilayer, glycoproteins, and proteins. Circulating proteins interact with the carbohydrate moieties of the glycoproteins. Several surface receptors are known to exist. Some of the more common receptors bind thrombin, adenosine diphosphate (ADP), TXA2, fibrinogen, collagen, and vWF.6 Platelets contain three types of storage granules: (1) dense granules, which contain serotonin, ADP, adenosine triphosphate (ATP), and calcium; (2) α-granules, which contain coagulation proteins (high-molecular-weight kininogen [HMWK], fibrinogen, fibronectin, factor V, vWF, platelet factor 4), growth factors, and adhesion proteins (fibronectin, thrombospondin, P-selectin); and (3) lysosomes.
Platelet activation is associated with numerous downstream signals, including protein kinase C activation, inositol triphosphate formation, intracellular calcium mobilization, and generation of arachidonic acid. Arachidonic acid is then converted by cyclooxygenase-1 (COX-1) to prostaglandin endoperoxides (PGG2, PGH2). PGG2 is converted to TXA2 by thromboxane synthetase. PGG2, PGH2, and TXA2 stimulate further aggregation and platelet granule release.7
Regardless of the agonist, the final common pathway for platelet aggregation involves a conformational change in the GP IIb-IIIa complex that leads to the reversible exposure of binding sites for fibrinogen, which allows fibrinogen to form bridges between adjacent platelets.8
Numerous medications inhibit platelet function at several steps in the pathway above. Aspirin irreversibly inhibits platelet COX-1, inhibiting TXA2-mediated platelet aggregation for the life of the platelet. Ticlopidine and clopidogrel inhibit ADP-mediated platelet activation and aggregation.9,10 Novel GP IIb-IIIa inhibitors prevent platelet aggregation by blocking the binding of fibrinogen.11
Coagulation Activation
The sequence of enzymatic events leading to thrombin formation is the coagulation cascade (see Figure 5-1). The intrinsic pathway is activated when plasma is exposed to a negatively charged surface such as subendothelium, collagen, or endotoxin. Factor XII is activated to XIIa by the interaction of HMWK, prekallikrein, and the negatively charged surface; however, the physiologic significance of factor XII activation is unclear because deficiencies in factor XII, HMWK, and prekallikrein are not associated with any clinical bleeding diatheses.
The extrinsic pathway is the more physiologic route for the generation of thrombin and fibrin. It is initiated by the exposure of tissue factor, which binds to low levels of circulating factor VIIa in the presence of calcium (TF-VIIa).12,13 This complex activates factor X and factor IX.14 Factor Xa alone does not generate thrombin efficiently; however, factors Xa and thrombin together can activate factors VII, V, and VIII. Factors Va and VIIIa are critical components of the prothrombinase and tenase complexes (see Figure 5-1), respectively, which are 105-fold to 106-fold more active at generating thrombin than their serine protease factors acting independently.13
Thrombin proteolytically cleaves peptides from the fibrinogen molecule, resulting in the polymerization of fibrin monomers to form a gel. Thrombin also activates factor XIII in a reaction that is greatly accelerated (>80-fold) by the presence of fibrin.15 Factor XIIIa covalently cross-links adjacent fibrin monomers, forming a stable clot that is more resistant to lysis by plasmin.
Coagulation Inhibition
Several mechanisms have evolved to control the rate of thrombin and fibrin formation (Figure 5-2). Antithrombin is a serine protease inhibitor that is synthesized in the liver and endothelial cells. AT inhibits numerous coagulation factors, but its most important targets are thrombin and factor Xa. AT activity is enhanced at least 1000-fold whenever it binds to circulating heparin or endothelial-bound heparin-like molecules. After the AT–heparin complex binds to an activated coagulation factor, the heparin dissociates and continues to act as a catalyst for the formation of other AT–serine enzyme complexes.
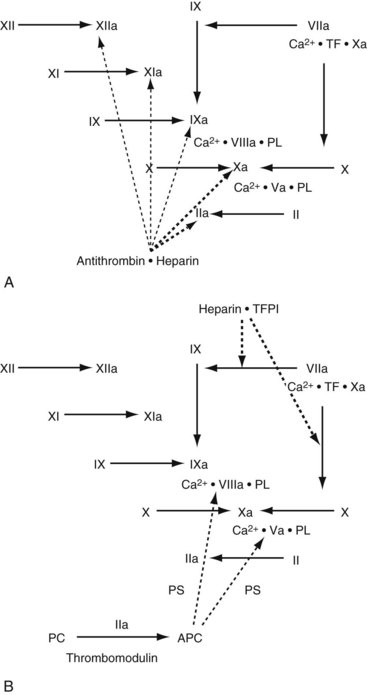
TFPI is an enzyme inhibitor synthesized by the endothelium and megakaryocytes.16,17 It binds to the TF-VIIa-Xa complex and inhibits the further activation of factors X and IX.18 TFPI is constitutively expressed on the endothelium, and its activity and antigen levels increase dramatically after the administration of heparin.
Thrombomodulin (TM) is a proteoglycan expressed on the surface of most endothelial cells2 that binds thrombin, causes a conformational change in the substrate binding site, and renders the thrombin molecule incapable of binding active coagulation factors. TM also accelerates the inactivation of thrombin by AT.19,20
Protein C and protein S are synthesized by the liver, but protein S has also been found in endothelium and platelets.21,22 Activated protein C binds to protein S on the endothelial or platelet surface and cleaves several peptide bonds in factors Va and VIIIa, resulting in decreased formation of the prothrombinase and tenase complexes.
Fibrinolysis.
Plasminogen, an inactive precursor synthesized in the liver, can be converted to plasmin by several plasminogen activators. Circulating t-PA does not activate plasminogen efficiently; however, both t-PA and plasminogen have high affinity for fibrin, which acts as a template for accelerated plasminogen activation (>1000-fold).23,24 Thus, the primary role for t-PA–activated plasmin is the formation of fibrin degradation products. Alternatively, exogenously administered t-PA may also activate plasminogen, which is bound to one of the fibrin degradation by-products, resulting in the release of free plasmin.25,26 This release can lead to the limited breakdown of fibrinogen, factor V, and factor VIII and to a systemic fibrinolytic state.
Three types of urokinase plasminogen activator (u-PA) have been studied. The precursor, pro-urokinase (single-chain u-PA), has a low level of enzymatic activity and no affinity for fibrin, but it demonstrates specificity against fibrin-bound plasminogen. Single-chain u-PA is readily converted by plasmin or kallikrein to the more active two-chain u-PA, which has a high-molecular-weight and a low-molecular-weight form. Commercially produced urokinase is composed primarily of the low-molecular-weight variant. Two-chain u-PA activates circulating plasminogen and fibrin-bound plasminogen equally well, resulting in a more pronounced systemic fibrinolysis.27
Preoperative Evaluation
Laboratory Screening
Platelet count is a key component in evaluating the patient with suspected thrombocytopenia; however, this test does not offer information regarding platelet function. Platelet function analyzers (PFA-100) are used to quantify congenital and acquired platelet dysfunction and von Willebrand disease. They are also helpful when preoperatively screening those with a positive family history of bleeding disorders, or those with liver or renal disease. The PFA-100 can be used to identify possible causes for intraoperative or postoperative bleeding, monitor treatment for von Willebrand disease, and identify those high-risk patients resistant to aspirin therapy. Essentially the PFA-100 measures how fast platelets adhere, activate, and aggregate into a platelet plug onto a collagen coated membrane in the presence of either epinephrine or ADP. In the presence of epinephrine, the cartridge (CEPI) allows for the detection of aspirin-induced defects and in the presence of ADP, the cartridge (CADP) allows for detection of more severe platelet defects.28
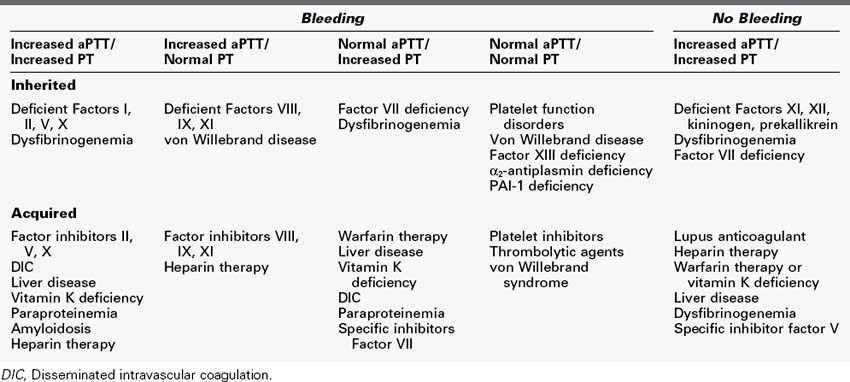
In those patients with elevated aPTT, the presence of platelet inhibitors may be suspected. A useful test in determining the presence of platelet inhibitors is the mixing study. In a mixing study, normal plasma is added in a 1 : 1 ratio to a patient’s plasma. If the aPTT corrects to normal, a specific factor deficiency (factors VIII, IX) is suspected. If the aPTT does not correct with the addition of normal plasma to the sample, the test is suggestive of the presence of a specific or nonspecific factor inhibitor in the patient’s sample. The abnormal mix then can be incubated at 37°C for 30 to 60 min and reassessed. If there is no change in the aPTT, the patient likely has a nonspecific inhibitor such as lupus anticoagulant. If the aPTT increases after incubation, the patient likely has a specific factor inhibitor such as anti–factor VIII antibodies. Mixing studies are sensitive but not specific and should be used only as a screening test. If lupus anticoagulant or other factor inhibitors are suspected, further testing is required to confirm the diagnosis.28 The common causes of elevated PT and aPTT are shown in Table 5-1.
Platelet Disorders
Thrombocytopenia
Thrombocytopenia can occur from increased platelet destruction, abnormal production, dilution, or temporary sequestration (usually in the spleen). Increased destruction can occur via nonimmune or immune mechanisms. Non–immune-mediated thrombocytopenia occurs in hemolytic-uremic syndrome, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation (DIC), and some vasculitides. In these syndromes, platelets are stimulated to aggregate within the microcirculation, often affecting the brain, kidneys, heart, lungs, and adrenal glands.29 Early plasmapheresis and plasma transfusion (platelet-poor fresh frozen plasma [FFP], cryoprecipitate-poor plasma), along with high-dose glucocorticoid administration, can reverse most cases of thrombotic thrombocytopenic purpura.30,31 Platelet transfusions should be used only for intracerebral or other life-threatening hemorrhagic complications. The treatment for hemolytic-uremic syndrome varies considerably but may include hemodialysis, heparin therapy, and plasma exchange, depending on the duration and severity of the illness. GP IIb-IIIa inhibitors may become a useful adjunct in hemolytic-uremic syndrome.32
Thrombocytopenia commonly occurs after massive transfusions of banked blood. Only 10% of platelets remain viable in blood held in cold storage for longer than 24 hours. In general, the replacement of one blood volume decreases the platelet count by one third to half.33 Nevertheless, abnormal bleeding is uncommon, and the routine administration of platelets following massive transfusion is not warranted unless hemorrhage is ongoing.34 Hypothermia (body temperature less than 32°C) also may cause thrombocytopenia, but the mechanism remains unclear, although it is known that sequestration of platelets during hypothermia occurs. Platelets appear to activate, release α-granule products, aggregate, and sequester in the portal circulation. Rewarming can cause a significant portion to return to the circulation. Cold-induced coagulopathy is best prevented by transfusing warmed blood products and maintaining the core body temperature greater than 32°C.
Qualitative Disorders of Platelet Function
Von Willebrand disease is the most common inherited bleeding disorder, characterized by a deficiency or defect in vWF. It has been classified into six subtypes (1, 2A, 2B, 2M, 2N, 3), with type 1 being the most common (70%).35 Type 1 von Willebrand disease is usually transmitted as an autosomal dominant trait with incomplete penetrance. In general, patients manifest epistaxis, ecchymoses, menorrhagia, and posttraumatic or postsurgical bleeding. Decreased platelet adherence causes prolongation of the bleeding time. The aPTT also may be elevated, because most patients with this disease have concomitant decreases in factor VIII coagulation activity (VIII:C). Ristocetin agglutination of platelets is impaired, but can be corrected with the addition of vWF-rich cryoprecipitate.
Treatment of von Willebrand disease can consist of replacement (cryoprecipitate, purified factor VIII concentrates, platelet transfusions) or nonreplacement (vasopressin, antifibrinolytic agents) therapy. Approximately 80% of patients with type 1 disease respond to desmopressin acetate (1-deamino-8-D-arginine vassopressin, DDAVP) with increased vWF:Ag and VIII:C (within 60 minutes), which can last for 4 to 6 hours. Unfortunately, response to therapy cannot be predicted without trial administration. Repeated administration of DDAVP (every 12 hours) may be required in patients with type 1 disease who undergo surgical procedures. Most type 2 and type 3 patients do not respond to DDAVP. Antifibrinolytic agents (ε-aminocaproic acid, tranexamic acid) have been used for the treatment of mucocutaneous bleeding and for prophylaxis during oral surgical procedures.36 Patients who are unresponsive to DDAVP may require replacement therapy during the perioperative period. Until recently, cryoprecipitate (rich in vWF, factors VIII and XIII, and fibronectin) was the treatment of choice. More recently, some purified factor VIII concentrates, which contain large quantities of multimeric vWF, and a newly formulated vWF concentrate have been used successfully.37 There are no clear guidelines regarding the amount and frequency of administration; replacement therapy is largely empirical. The bleeding time and factor VIII levels are used to monitor response to replacement therapy.
Storage pool diseases are a group of rare hereditary disorders characterized by deficiencies in platelet granules, their contents, or both. These deficiencies include α-granule contents (gray platelet syndrome), δ-granule storage diseases (Wiskott-Aldrich syndrome, Hermansky-Pudlak syndrome, Chédiak-Higashi syndrome), and αδ-granule storage diseases.38 Cryoprecipitate and platelet transfusions can be used in the perioperative period. DDAVP also has been used to decrease the requirement for transfusions.
Acquired qualitative platelet abnormalities can be caused by certain drugs, uremia, cirrhosis, myeloproliferative disorders, and dysproteinemias. Aspirin irreversibly acetylates platelet cyclooxygenase-1, inhibiting thromboxane- and endoperoxide-mediated platelet activation for the life of the platelet. The effect of aspirin on the bleeding time is variable and may depend largely on the technique used to perform the test.39,40 Nonsteroidal antiinflammatory drugs (e.g., indomethacin, phenylbutazone, ibuprofen) reversibly inhibit cyclooxygenase. Numerous antibiotics, including some β-lactams, cephalosporins, and nitrofurantoin, impair platelet aggregation and prolong the bleeding time. Mechanisms can include inhibition of agonist binding to the membrane receptor and inhibition of intracellular signal transduction. Platelet GP IIb-IIIa inhibitors (e.g., abciximab, eptifibatide, tirofiban) block the binding of fibrinogen to the GP IIb-IIIa receptor and effectively prevent platelet aggregation in a dose-dependent fashion. Correction of bleeding can be accomplished with platelet transfusions.
Uremia causes defective platelet adherence and aggregation, resulting in a prolonged bleeding time. Clinical manifestations can include petechiae, ecchymoses, and mucocutaneous bleeding. The pathophysiology remains unclear, but may involve impaired thromboxane and calcium metabolism or defective platelet-subendothelial adhesion (via vWF). DDAVP has been shown to shorten bleeding times preoperatively in uremic patients.41 Intravenous DDAVP, 0.3 to 0.4 µg/kg over 15 to 30 minutes, shortens the bleeding time in most patients within 1 hour. Hemodialysis, peritoneal dialysis, and infusions of cryoprecipitate and conjugated estrogens have been used with some success.42
Disorders of Secondary Hemostasis
Congenital Disorders
Patients with hemophilia A who require major surgery should receive factor VIII replacement to achieve 100% of normal activity just before the procedure. For each unit per kilogram of body weight infused, the factor VIII level is increased by approximately 0.02 U/mL (normal activity is 1 U/mL).43 Levels should be monitored postoperatively, and replacement therapy should be repeated every 12 hours to maintain at least 50% of normal activity until all wounds are healed.44 Factor VIII levels can be restored using donor-directed cryoprecipitate, virus-inactivated factor VIII concentrate, or recombinant factor VIII. DDAVP (which increases factor VIII levels) and ε-aminocaproic acid may be used as adjunctive therapies in patients with mild hemophilia to reduce or avoid the need for replacement therapy during oral or minor surgical procedures.
Rare coagulation factor deficiencies of factors II, V, VII, and X occur with a prevalence of 1 : 500,000 to 1 : 1,000,000. They are usually transmitted with an autosomal recessive pattern. The most severe complications occur with deficiencies of factors II and X.45 In general, only low levels of factor activity (10% to 20% of normal) are required for normal hemostasis. Replacement therapy for factors II and X can be accomplished with fresh frozen plasma or factor concentrates. Factor IX concentrates contain significant amounts of factors II and X and can be used for their replacement. The short half-life of factor VII requires a more frequent replacement schedule using factor VII concentrates. Recombinant factor VIIa also can be used for factor VII deficiencies. Factor V deficiencies can be treated with fresh frozen plasma, because factor V concentrates are not yet commercially available.
Abnormalities of fibrinogen and fibrinolysis are also heritable. Afibrinogenemia is a rare disorder transmitted as an autosomal recessive trait; hypofibrinogenemia can occur in heterozygous individuals. Clinical manifestations include gastrointestinal and mucous membrane bleeding, hemarthroses, intracranial hemorrhage, and recurrent fetal loss. The PT and aPTT, which are markedly prolonged, usually correct when mixed with normal plasma. Replacement therapy with cryoprecipitate is usually reserved for active bleeding, the perioperative period, and prophylaxis during pregnancy. The level of fibrinogen necessary for hemostasis ranges between 50 and 100 mg/dL. Each unit of cryoprecipitate usually increases the fibrinogen level by approximately 10 mg/dL.46
Dysfibrinogenemias are a heterogeneous group of disorders that can cause defective fibrin formation, polymerization, cross-linkage, or impaired fibrinolysis. Patients may manifest mild to moderate bleeding diatheses (30%) or recurrent thromboses (20%).47 The PT and aPTT usually are prolonged. Functional assays for fibrinogen are abnormal, whereas antigenic assays are normal. Cryoprecipitate is indicated for hemorrhage, but contraindicated for acute thrombotic episodes.
Congenital hyperfibrinolytic states can result in delayed bleeding. The congenital hyperfibrinolytic states include heterozygous and homozygous α2-antiplasmin deficiencies and functionally abnormal or deficient PAI-1.48 The whole blood clot lysis time and the euglobulin clot lysis time are characteristically shortened. Antifibrinolytic agents (ε-aminocaproic acid or tranexamic acid) are recommended for the management of active bleeding.49
Acquired Disorders
DIC is characterized by the systemic generation of fibrin, often resulting in the thrombosis of small- and medium-sized blood vessels. The consumption of clotting factors and platelets also results in impaired coagulation and hemorrhagic complications. DIC is mediated by several cytokines (including tumor necrosis factor-α and interleukin-6), which result in the systemic generation of TF, thrombin, and fibrin.50 Fibrinolytic activity, which is initially increased via the release of t-PA, becomes depressed in response to elevated PAI-1.50,51 DIC can develop in association with bacterial infections (gram-positive and gram-negative infections), trauma, malignancy, obstetric complications, hemolytic transfusion reactions, giant hemangiomas (Kasabach-Merritt syndrome), and aortic aneurysms. A compensated DIC (present in more than 80% of patients who undergo major surgery), in which coagulation factors and platelets are replaced as they are consumed, may be asymptomatic or may appear with ecchymoses and petechiae. Surgery, trauma, hypotension, or transfusion reactions can exacerbate the coagulopathy and hypofibrinolysis, leading to excessive bleeding and intravascular thrombosis.
A combination of laboratory tests may help to confirm the clinical diagnosis of DIC. These tests include detection of thrombocytopenia or a rapidly decreasing platelet count, prolongation of the PT and aPTT, and the presence of fibrin degradation products (d-dimer assay, latex agglutination for fibrous degradation products). Extrinsic pathway coagulation proteins (factors II, V, VII, and X) and physiologic coagulation inhibitors (AT, protein C) usually are depressed, whereas vWF and factor VIII levels may be increased.52 The fibrinogen level is variably affected by DIC.
The first goal of management is elimination of the cause of DIC. When this is possible, the intravascular coagulation ceases with the return of normal hemostasis. In severe DIC, with ongoing blood loss, patients are best managed by replacing deficient blood elements using fresh frozen plasma (up to 6 units per 24 hours) and platelets while the precipitating cause of DIC is eliminated.50 Administration of AT and protein C concentrates may retard the consumption of coagulation factors, although this remains to be proved. Some trials have demonstrated a benefit with the administration of heparin or low-molecular-weight heparin (LMWH).53,54 Given that patients with DIC already have a coagulopathy, heparin should be used cautiously (lower IV doses of 300 to 500 units/hour) and with careful clinical observation and laboratory monitoring. Direct thrombin inhibitors (hirudin, recombinant TM), activated protein C, and extrinsic pathway inhibitors (recombinant TFPI) are under investigation as well.
Management of Anticoagulation
Active Hemorrhage
Controlled clinical studies have shown that treatment with vitamin K antagonists (VKAs) increases the risk of major bleeding by 0.5% per year and the risk of intracranial hemorrhage by 0.2% per year.55 Risk factors associated with hemorrhage in patients treated with VKAs include target international normalized ratio (INR) greater than 3,56 patient age, cytochrome P450 CYP2C9 polymorphisms that decrease VKA metabolism,57 and renal and hepatic insufficiency. The addition of antiplatelet therapy and nonsteroidal antiinflammatory medications in the setting of VKA therapy also increases the risk of major bleeding 2.5-fold greater than normal and increases the risk of gastrointestinal bleeding 11-fold, respectively.58–60
In the face of bleeding, the reversal of VKA therapy is critical and varies depending on the INR and clinical status of the patient. To reverse VKA therapy, vitamin K replacement is often the first line of therapy in clinically stable patients with minimum signs of hemorrhage. Vitamin K replacement can be administered orally, but the INR usually takes 24 hours to normalize in this case. IV vitamin K can normalize INR within 12 to 16 hours. Intramuscular and subcutaneous routes should be avoided because absorption can be unpredictable and delayed.61 The recommended dose for vitamin K replacement varies depending on the INR. If INR is less than 7, 2.5 to 5 mg of vitamin K is effective. If the INR is greater than 7, 5 to 10 mg of vitamin K is required.
Factor replacement including the use of FFP, prothrombin complex concentrates (PCCs), and recombinant factor VIIa can be considered when active hemorrhage is apparent and rapid correction of INR is necessitated. When administered, FFP elicits a 2% to 4% rise in factor activity per unit infused. Large volumes of FFP are often required to correct an elevated INR, and there is an associated risk of transfusion-related acute lung injury (TRALI), anaphylaxis, and transmission of viral infections with FFP administration. In addition, FFP must be thawed before infusion and needs to be crossmatched to assure ABO compatibility limiting the rapidity of INR correction and hemorrhage cessation. Because of the variability of vitamin K–dependent clotting factors in FFP, others have studied the effect of clotting factor concentrates on the INRs of anticoagulated patients who require rapid correction. Makris and colleagues62 found that patients given FFP did not normalize their INR, whereas those treated with clotting factor concentrates did, largely in part to increased factor IX levels in the clotting factor concentrate compared to the factor IX levels found in FFP.
Recombinant factor VII (rVIIa) replacement can also be used to correct INR and halt hemorrhage in patients taking VKAs. The rVIIa contains hamster proteins, bovine IgG, and mouse IgG. The mechanism of action for rVIIa is that it complexes with tissue factor to activate factors IX and X, initiating the clotting cascade while bypassing the activation of factors VIII and IX. A typical dose of rVIIa is 5 to 16 µg/kg IV.63 A comparison of rVIIa to PCC (Octaplex, Octapharma AG, Lachen, Switzerland) showed that although both rVIIa and PCC corrected INR, only PCC restored endogenous thrombin generation, which is the key endpoint in restoring hemostasis.64
PCCs are lyophilized concentrates of a standardized amount of factor IX and different amounts of factors II, VII, and X that vary by manufacturer. PCCs are currently approved in Europe, Australia, and Canada for use in patients with factor IX deficiency or for rapid reversal of VKA therapy, whereas PCCs are approved by the U.S. Food and Drug Administration (FDA) only for factor IX deficiency. Although many PCCs are poor in factor VII, they are virally inactivated, undergo prion reducing processes, are lyophilized rather than frozen, and are administered in small volumes (40 to 80 mL). Should PCCs be approved by the FDA for use in patients taking VKAs, it should be noted that the use of PCCs is indicated specifically for those patients exhibiting major bleeding or requiring urgent surgical procedures (<6 hours). PCCs are not recommended for VKA reversal in the elective setting, for elevated INR without bleeding or an urgent need for surgery, for use in massive transfusions, coagulopathy of liver disease, patients with a recent history of thrombosis, ischemic stroke or DIC, or patients with a history of heparin-induced thrombocytopenia. Complications of PCC use include thrombosis, DIC, hemorrhage, and viral transmission.65
Perioperative Management
Patients who are being treated with VKAs before surgery must be assessed to determine the best mode of anticoagulation management for that patient to decrease the risk of bleeding and thrombosis. The general options for managing these patients in the perioperative period include continuing anticoagulation, decreasing warfarin dosage, or discontinuing warfarin and administering bridging therapy with LMWH or unfractionated heparin using the American College of Chest Physicians (ACCP) guidelines published in 2008.66 Currently, the ACCP guidelines are most frequently referred to as the standard of care for these complex patients; however, there is some evidence supporting the continuation of anticoagulation therapy or decreasing the dose perioperatively to reduce the risk of bleeding while maintaining adequate anticoagulation to prevent arterial thrombosis.
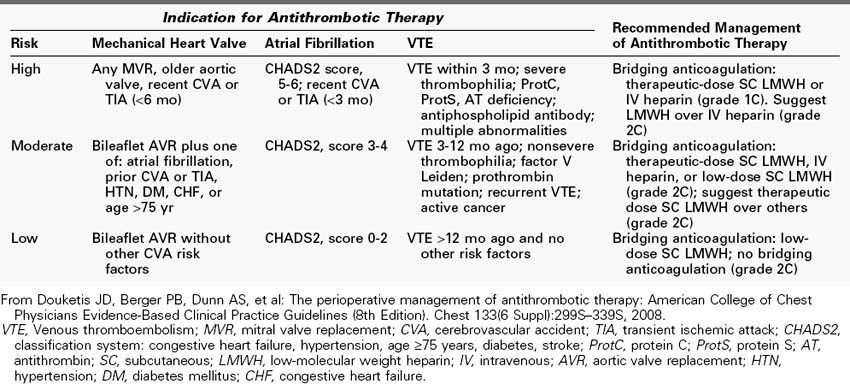
Table 5-2 shows the risk stratification for patients receiving anticoagulation therapy for three common indications, including mechanical heart valves, atrial fibrillation, and venous thromboembolism. Table 5-2 also shows the recommended management of anticoagulation therapy by risk stratification. Generally, in patients deemed to be at high risk, bridging anticoagulation with therapeutic-dose LMWH or IV heparin is recommended (grade 1C) and LMWH is recommended over IV heparin (grade 2C).
TABLE 5-2 Risk Stratification and Recommendations for Perioperative Arterial or Venous Thromboembolism
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


