Hematologic Aspects of Pediatric and Adolescent Heart Disease: Bleeding, Clotting, and Blood Component Abnormalities
Therese M. Giglia
Char Witmer
The cardiovascular system is both the conduit and the propeller of the circulating blood, and as such, a mandatory codependence exists between these two organ systems. As expected, perturbations in one system result in alterations in the other and vice versa. The purpose of this chapter is to describe the cardiovascular effects of hematologic derangements on the normal heart as well as the hematologic problems seen in children and adolescents with congenital and acquired heart disease. The chapter begins with an overview of basic principles of hematology in the developing child and progresses to discussions of abnormalities in individual blood components and bleeding and how each affects the normal heart as well as the heart of the child and adolescent with congenital and acquired heart disease. Since thrombosis is becoming more commonly recognized as a major source of morbidity and at times mortality in children with heart disease, the chapter ends with a detailed discussion of thrombosis in pediatric heart disease including a description of anticoagulants, antiplatelet agents, and thrombolytic therapy commonly used in children with these disorders.
Basic Principles of Hematology
Red Blood Cells
Red blood cells (RBC) are nonnucleated biconcave discs whose major cellular component is hemoglobin (Hb), an oxygen transport protein. Erythropoietin, a hematopoietic growth factor produced in the kidney, is the major regulator of red cell production. A normal red cell life span is approximately 120 days. Normal developmental factors influence Hb values in children including age, gender, and sexual maturity necessitating the need for age-appropriate reference values.
Hb production begins early in gestation and undergoes a series of transitions. Hb is formed by two pairs of identical subunits called globin chains. Fetal erythropoiesis consists of an orderly evolution through a series of different Hbs. All forms of Hb are made up of a combination of two α-like globin proteins (α or ξ) and two β-like globin proteins (β, δ, γ, or ε). In the embryo, the predominant Hbs include Gower 1 (ξ2ε2), Gower 2 (α2ε2), and Portland (ξ2γ2). In the fetus, there is a transition to fetal Hb (α2γ2). After birth, there is a final switch to adult Hb (α2β2).
At birth, neonates have a mean Hb of 15.9 g/dL (±1.86) and an elevated mean corpuscular volume (MCV) of 110 fl (±5) (1). After birth, red cell production quickly decreases likely secondary to the abrupt increase in oxygen concentration. The half-life of neonatal red cells is shorter (average 23.3 days) than in adults and is even shorter in premature infants (average 16.6 days) (2). The Hb naturally decreases over the first 2 to 3 months of life (physiologic nadir) and then slowly increases in the fourth to sixth months of life. Throughout childhood, there is a mild increase in the mean Hb. In males during puberty, as the Tanner stage increases, the Hb increases. There is no relationship between Hb and Tanner stage in females.
White Blood Cells
White blood cells (WBC) play an integral role in the immune system. There are five types of WBC including neutrophils, eosinophils, basophils, lymphocytes, and macrophages/monocytes. Individual WBC percentages have limited clinical utility; instead, absolute counts should always be considered. The WBC reference range will vary with age. In general, newborns have a higher total WBC (mean 18.1 k/μL) that will quickly decrease over the first week of life. Lymphocyte predominance is seen from 2 weeks to approximately 5 years of age, and then neutrophils become predominant.
Hemostasis
Platelets are small anucleated cell particles that are made in the bone marrow via fragmentation of megakaryocytes; production is mediated via thrombopoietin. Platelets circulate for approximately 7 to 10 days and are subsequently removed via the reticuloendothelial system. By 18 weeks of gestation, the plasma platelet concentration reaches the adult range of 150 to 450 k/μL. At birth, neonates have the same platelet count range and volume as adults. Data regarding platelet function in neonates demonstrate hyporeactivity to some agonists and hyperreactivity to others (1). Platelets play an integral role in hemostasis.
Hemostasis refers to the coordinated process that stops bleeding at the site of vascular injury through the formation of an impermeable platelet and fibrin plug. Hemostasis is achieved through the following three main mechanisms:
Vascular constriction
Primary platelet plug formation (primary hemostasis)
Clot propagation through fibrin formation (secondary hemostasis)
Vascular constriction decreases blood flow at the site of injury. Platelets adhere to the exposed subendothelium, through von Willebrand factor (vWF) tethering, forming an initial platelet plug. Simultaneously, coagulation is initiated by the exposure of tissue factor. Tissue factor binds and activates FVII that activates the coagulation cascade, resulting in a small thrombin burst. This small thrombin burst stimulates further platelet activation and the activation of coagulation on the platelet surface. On this increased platelet surface, a large amount of thrombin is formed that is sufficient to convert fibrinogen to fibrin
leading to stable clot formation. To limit clot formation at the site of injury, activated procoagulant proteins are inhibited by anticoagulant proteins (protein C, S, antithrombin, thrombomodulin, heparin cofactor II, and tissue factor pathway inhibitor). Clot degradation is initiated by the fibrinolytic system (plasminogen, tissue plasminogen activator [tPA], and urokinase plasminogen activator). This is a finely balanced system, and a derangement at any level can result in a tendency for bleeding or a prothrombotic state.
leading to stable clot formation. To limit clot formation at the site of injury, activated procoagulant proteins are inhibited by anticoagulant proteins (protein C, S, antithrombin, thrombomodulin, heparin cofactor II, and tissue factor pathway inhibitor). Clot degradation is initiated by the fibrinolytic system (plasminogen, tissue plasminogen activator [tPA], and urokinase plasminogen activator). This is a finely balanced system, and a derangement at any level can result in a tendency for bleeding or a prothrombotic state.
In the neonate, these hemostatic processes are in place but in different concentrations than adults. In normal postnatal development, many values normalize by 6 months of age, although changes can still be seen throughout childhood (3,4). Understanding the difference in neonatal values is imperative when interpreting coagulation studies to ensure the correct diagnosis of either a bleeding or clotting disorder. It also has direct implications for the use of specific hemostatic interventions in a neonate (i.e., unfractionated or low–molecular-weight heparin [LMWH] therapy).
Coagulation proteins do not cross the placenta and are independently synthesized by the fetus; most are present by 10 weeks of gestation and gradually increase with gestational age (5,6). In a neonate, the following procoagulant proteins are decreased including the vitamin K–dependent factors (II, VII, IX, and X) and the contact pathway factors (XI, XII, prekallikrein, and high–molecular-weight kininogen) (4,6,7). This results in a prolonged prothrombin time (PT) and partial thromboplastin time (PTT) when compared to normal adult values. Conversely, neonates have increased plasma vWF levels and elevated levels of circulating ultra-large von Willebrand multimers (8,9). Similar to the procoagulant factors, the inhibitors of coagulation are also decreased. The anticoagulant proteins including protein C, S, antithrombin, heparin cofactor II, and tissue factor pathway inhibitor are decreased, resulting in a slower rate of thrombin inhibition (4,6,7,10). The fibrinolytic system is also depressed secondary to a unique neonatal glycoform of plasminogen that is inefficiently converted to plasmin (11). Neonates will have markedly elevated D-dimer values at birth lasting up to 3 days (7,10).
Hematologic Disorders
Special Consideration of Hematologic Disorders in Congenital and Acquired Heart Disease
Adolescents and children with congenital and acquired heart disease are at increased risk for hematologic abnormalities including red cell anomalies, bleeding, and thrombosis. The following sections discuss individual hematologic disorders describing the effects on the normal heart and in addition paying attention to particular concerns regarding the child and adolescent with congenital and acquired heart disease.
TABLE 75.1 Laboratory Evaluation for a Microcytic Anemia | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
Disorders of Red Blood Cells
Anemia
Anemia is defined as a decrease in hemoglobin (Hb) that is two standard deviations below the mean value for age. Physiologically, anemia can be divided into three main categories: decreased or ineffective RBC production, increased RBC destruction, or blood loss. The etiology of an anemia can be determined using a morphologic approach based on the red cell MCV. The initial approach to anemia should include a thorough history, physical examination, complete blood count (CBC) with differential, reticulocyte count, and review of the peripheral smear.
The differential for a microcytic anemia is rather narrow and includes acquired and congenital causes. The most common acquired cause in pediatrics is iron deficiency. Premature infants are at increased risk for iron deficiency secondary to decreased in utero iron absorption, decreased birth weight, and concurrent anemia. Toddlers commonly have dietary-induced iron deficiency when there is excess milk intake combined with poor solid food intake. The congenital causes for a microcytic anemia include β- or α-thalassemia trait, other forms of thalassemia, sickle cell combined with thalassemia, or anemia of chronic disease. Table 75.1 provides a summary of laboratory values to help differentiate between the common causes of a microcytic anemia. An Hb electrophoresis helps in diagnosing other forms of thalassemia and sickle cell disease (SCD).
A normocytic anemia has a much broader differential than a microcytic anemia. Figure 75.1 provides a flow diagram for the approach to a normocytic anemia. The reticulocyte count should be used to further classify the anemia into two broad categories of increased or decreased red cell turnover. The reticulocyte count needs to be adjusted for the degree of anemia to determine if it is truly elevated. A reticulocyte index (RI) can be calculated as:
An RI >3 indicates increased red cell turnover and an RI <3 indicates decreased red cell turnover. In patients with an elevated RI, it is important to differentiate between bleeding versus red cell destruction (hemolysis). Laboratory markers of hemolysis include
an increased lactate dehydrogenase (LDH), elevated indirect bilirubin, elevated aspartate aminotransferase (AST) with a concomitant normal alanine transaminase (ALT), or a decreased haptoglobin.
an increased lactate dehydrogenase (LDH), elevated indirect bilirubin, elevated aspartate aminotransferase (AST) with a concomitant normal alanine transaminase (ALT), or a decreased haptoglobin.
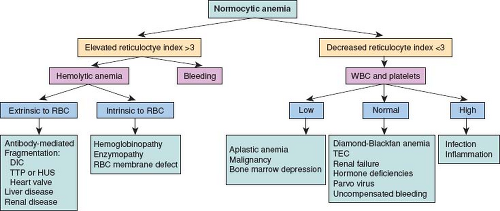 Figure 75.1 The differential for a normocytic anemia. DIC, disseminated intervascular coagulation; TTP, transient thrombocytopenic purpura; RBC, red blood cell; TEC, transient erythroblastopenia of childhood. |
Patients with congenital or acquired heart disease are at increased risk for developing an acquired hemolytic anemia from an increase in shear forces most commonly seen in patients with prosthetic valves. These patients will develop an anemia with an elevated RI. Review of the peripheral smear will show red cell fragments and they will have evidence of intravascular hemolysis with a decreased haptoglobin, increased LDH, and hemoglobinuria. Other markers of hemolysis including indirect bilirubin or AST may or may not be elevated.
Macrocytic anemia is much less common in pediatrics. The differential includes vitamin B12 or folate deficiency, hypothyroidism, bone marrow failure, significant reticulocytosis, liver disease, or medications. Common medications that can increase the MCV include hydroxyurea, zidovudine, or chemotherapeutic agents. In the neonatal period, infants normally have an increased MCV due to the high percentage of hemoglobin F. In Down syndrome, an increased MCV is common in up to two-thirds of patients (12).
Anemia and RBC Transfusion in Children and Adolescents with Congenital Heart Disease (CHD)
Since children and adolescents with cyanotic heart disease have higher Hb levels commensurate with their desaturation and higher than age-matched controls without cyanotic heart disease, assessment of anemia is more challenging. Recognition of this discrepancy is imperative to being able to diagnose anemia in patients with cyanotic CHD. Paying attention to a particular patient’s baseline values and a change over time will be more informative than age-matched normal values.
Children with cyanotic defects are commonly transfused to an Hb >14 g/dL especially postoperatively or during periods of hemodynamic instability to increase their oxygen-carrying capacity and optimize oxygen delivery. The rationale for this strategy is that a compromised cyanotic patient has limited ability to increase cardiac output to compensate for a low systemic oxygen delivery (13,14,15). Although a common practice, there is a paucity of data on the optimal Hb concentration and transfusion strategies in these patients. A single-center, prospective, randomized, controlled clinical trial compared a restrictive Hb strategy (mean Hb 11 g/dL, mean RBC transfusions 0.43) to a liberal strategy (mean Hb 13.9 g/dL, mean RBC transfusions 2.1) in children during the first 48 hours after bidirectional cavopulmonary or Fontan operation. No differences were found in mean or peak arterial lactate, arteriovenous or arterio-cerebral oxygen content, or clinical outcomes. A recent Cochrane Review on red cell transfusion for patients undergoing cardiac surgery for CHD, with or without cyanosis, was unable to assess this question adequately due to insufficient evidence (16). Further investigation is warranted.
Red Blood Cell Disorders with Particular Cardiac Manifestations
SCD and thalassemia are two congenital red cell disorders with well-defined cardiovascular complications. SCD is a group of inherited hemoglobinopathies that are the result of abnormal Hb production. It is a multisystem disease characterized by a chronic hemolytic anemia and vaso-occlusive complications resulting in episodes of acute illness and a chronic progression to end-organ damage. SCD is an autosomal recessive condition. The most common form of SCD is homozygous SS. Other variants of SCD are the result of other Hb variants combined with Hb S (i.e., SC, S-β0 thalassemia, or S-β+ thalassemia). More than 100,000 Americans are affected with SCD. The carrier rate in African Americans is approximately 8% (1 in 12).
In Hb S, an amino acid substitution in the β-globin gene from glutamic acid to valine ultimately leads to the polymerization of Hb S molecules, causing the red cell “sickling” effect with resultant vascular occlusion and hemolytic anemia. SCD was once seen as a disease where the morbidity and mortality were directly related to vascular occlusion but it is now evident that chronic hemolysis significantly contributes to the development of endothelial dysfunction and vasculopathy. With intravascular hemolysis, there is a release of free Hb that generates reactive oxygen species that are potent scavengers of nitric oxide (17). There is also a release of arginase that decreases nitric oxide bioavailability. Nitric oxide has several key roles in endothelial function including as a regulator of vasodilator tone and inhibitor of platelet and hemostatic activation (18,19,20). SCD confers a state of nitric oxide resistance, and human and animal studies have provided evidence that a reduction in nitric oxide is associated with vasoconstriction, decreased blood flow, platelet activation, and end-organ injury (21).
Cardiac involvement in SCD is quite common and is primarily the result of a chronic anemia with a compensatory increased cardiac output. In general, the heart is usually enlarged and a systolic ejection murmur is found in most patients. Fifty percent of pediatric patients will have electrocardiographic findings of left ventricular (LV) hypertrophy (22). The steady-state blood pressure in patients
with SCD is lower than that in age-, race-, and gender-matched controls (23). This finding is likely related to the steady-state anemia as well as renal losses of sodium and water. While vaso-occlusion is a common occurrence in SCD, myocardial infarction is quite rare although infarction of LV papillary muscles with mitral regurgitation has been reported (22,24,25). Autopsy studies show that atherosclerosis is also uncommon in this patient population (24).
with SCD is lower than that in age-, race-, and gender-matched controls (23). This finding is likely related to the steady-state anemia as well as renal losses of sodium and water. While vaso-occlusion is a common occurrence in SCD, myocardial infarction is quite rare although infarction of LV papillary muscles with mitral regurgitation has been reported (22,24,25). Autopsy studies show that atherosclerosis is also uncommon in this patient population (24).
Pulmonary hypertension (PH) is a common complication in adult patients with SCD with a reported prevalence of 6% to 32% and has been associated with significant morbidity and mortality (26,27). Studies with a higher prevalence of PH only used echocardiographic assessments of pulmonary artery systolic pressure with a cutoff tricuspid regurgitant (TR) jet velocity of 2.5 m/s. A prospective study in adult patients with SCD utilized right heart catheterization to confirm the echocardiographic findings and they reported a prevalence of elevated TR jet velocity of 27% (consistent with prior studies) but right heart catheterization confirmed PH in only 6% of subjects (27). Additionally, it is important to note that transient elevations in the TR jet velocity can be seen during vaso-occlusive episodes (28). Studies addressing PH in pediatric patients with SCD are limited but have not demonstrated the increased morbidity and mortality seen in adults (29,30,31,32,33,34). The prevalence of elevated TR velocities is similar in children as in adults; however, the significance of the findings and implications for prognosis remain unclear. Further pediatric studies that follow patients into adulthood are required. The 2014 Evidence-Based Management of Sickle Cell Disease Expert Panel Report from the National Institutes of Health where unable to make a recommendation for or against screening for PH in patients with SCD due to insufficient evidence (35). They only recommend evaluation in those patients with symptoms concerning for PH (35).
The etiology of PH in patients with SCD is likely multifactorial. Hemolysis is believed to play a key role in the development of PH through endothelial dysfunction. Other chronic hemolytic disorders have an increased incidence of PH including thalassemia, hereditary spherocytosis, and paroxysmal nocturnal hemoglobinuria. Other contributing factors in SCD may include chronic hypoxia, chronic thromboembolism, in situ thrombosis, parenchymal and vascular injury from sequestration of sickled erythrocytes, chronic liver disease, iron overload, and asplenia (36).
Patients with SCD who undergo surgery are at increased risk for morbidity and mortality (37,38,39). Specific morbidities include acute chest syndrome, cerebrovascular accidents, and infections. These risks are likely secondary to the underlying anemia from SCD, a propensity for microvasculature occlusion from sickled red cells, and asplenia. Improved outcomes have been reported in patients who received preoperative transfusions to a goal Hb level of 10 g/dL (which decreases the percent S and improves the patient’s baseline anemia), hydration, and aggressive intra- and postoperative monitoring (40,41). The patient’s hemodynamic status, that is, the effect of volume overload on a left-to-right shunt or ventricular dysfunction, will need to be considered. If a patient with SCD requires cardiac surgery or prolonged sedation for a cardiac procedure, a pediatric hematologist should be involved to work with the cardiologist/intensivist and cardiac anesthesiologist to determine the appropriate pre- and postoperative management.
Thalassemia syndromes are a group of inherited anemias resulting from defects in the production of Hb. Thalassemia is classified according to the globin chain that is under-produced, either α (α-thalassemia) or β (β-thalassemia) globin. A decrease in the production of either an α- or β-globin chain results in an excess of free globin chains that precipitate in the red cell and causes RBC membrane damage. The end result is anemia from RBC hemolysis and ineffective erythropoiesis in the bone marrow. α-Thalassemia is more commonly found in Southeast Asia, whereas β-thalassemia is more common in Mediterranean countries.
There are two α-globin genes located on chromosome 16. α-Thalassemias are usually the result of gene deletions. The severity of disease is related to the number of gene deletions present. One gene deletion is termed a silent carrier and has no hematologic manifestations. A two-gene deletion is termed α-thalassemia trait; the patient has a mild microcytic hypochromic anemia but is otherwise well with a normal Hb electrophoresis. A three-gene deletion results in Hb H disease. Hb H is composed of tetramers of β-globin. Patients with Hb H disease have a moderate microcytic hemolytic anemia and are not transfusion dependent. The Hb electrophoresis will demonstrate the presence of Hb H. The presence of a four-gene deletion is termed hydrops fetalis and results in severe anemia in the fetus with resultant intrauterine death without medical intervention.
There is one β-globin gene located on chromosome 11. Point mutations are the most common type of genetic mutation in β-thalassemia. β-Thalassemia trait occurs when only one gene is affected, resulting in a mild microcytic anemia. Hb electrophoresis shows increased Hb A2 and/or Hb F levels. In contrast, the inheritance of two affected β-globin genes results in a broad spectrum of clinical disease. Severity is determined by the residual amount of β-globin synthesis. The clinical phenotype ranges from transfusion dependence (β-thalassemia major) to a moderate anemia that does not necessitate chronic transfusions (β-thalassemia intermedia).
Treatment of β-thalassemia major consists of either life-long chronic red cell transfusions or bone marrow transplant. Chronic red cell transfusions correct the anemia and suppress ineffective erythropoiesis. Transfusions are life long and are typically given every 3 to 4 weeks with target nadir Hb of 9 to 10 g/dL. Unfortunately, a direct consequence of chronic transfusions is iron overload with excessive iron deposition in the liver, heart, and endocrine organs.
Currently, heart failure is the most common cause of death in patients with β-thalassemia major. Most of these deaths are attributable to cardiac iron overload (42,43). The most common cardiac abnormality in patients with heart failure from iron overload is a biventricular dilated cardiomyopathy, and severe right ventricular cardiomyopathy is evident in advanced disease (44). Iron deposition is greatest in the ventricular walls and less in the atria and the conduction system. Cardiomyocytes are also sensitive to oxidative damage from non–transferrin-bound iron. Pericarditis was common prior to the use of chelation therapy but appears to be decreasing in frequency (45). Paroxysmal atrial fibrillation is common and is typically associated with myocardial dysfunction; restoration of sinus rhythm does not usually reverse the cardiomyopathy (45). Currently, iron loading in the heart can be measured using MRI T2* technology, >20 ms indicates no significant iron loading, 10 to 19 ms indicates mild-to-moderate iron loading, and <10 ms indicates severe cardiac iron loading (46).
Iron overload is currently treated with chelation. First-line chelators currently in use in the United States include deferoxamine, which is given as a 12-hour infusion intravenously or subcutaneously or deferasirox, an oral medication given once daily. Deferiprone, another oral chelator, is approved as a second-line oral chelator in the United States. Variation may exist in organ-specific iron removal by chelator. Deferiprone appears to promote removal of cardiac iron over liver removal (47). If a patient has significant cardiac iron loading an escalation in chelation therapy is imperative. Escalation strategies including monotherapy with increased intensity or combination chelator therapy. Deferiprone in combination with an additional chelator may have improved cardiac iron removal versus monotherapy (48,49,50). Although a recent Cochrane review concluded that there are excessive toxicities with the use of deferiprone alone or in combination with other chelators without clear evidence of added benefit and suggested that further clinical trials are warranted (51). Reversal of the iron-induced cardiomyopathy is seen with intensive chelation (45).
Polycythemia
Polycythemia is defined as an increase in red cell volume; when the Hb or hematocrit is two standard deviations above the mean value
for age. Polycythemia can be classified by the response of erythroid progenitors to cytokines (i.e., erythropoietin) (52). In primary polycythemia, erythroid progenitors exhibit an excessive response to cytokines secondary to an inherited or acquired genetic mutation (i.e., polycythemia vera or erythropoietin receptor mutations). Secondary polycythemias are characterized by a normal response of the erythroid progenitors to elevated levels of cytokines. Secondary polycythemia is usually the result of a physiologic response to chronic hypoxia (i.e., cyanotic CHD or sleep apnea), autonomous erythropoietin production (tumor secretion), or exogenous erythropoietin administration. Rarely, secondary polycythemia can be due to Hb variants with an altered affinity for oxygen or genetic mutations that result in disordered hypoxic sensing.
for age. Polycythemia can be classified by the response of erythroid progenitors to cytokines (i.e., erythropoietin) (52). In primary polycythemia, erythroid progenitors exhibit an excessive response to cytokines secondary to an inherited or acquired genetic mutation (i.e., polycythemia vera or erythropoietin receptor mutations). Secondary polycythemias are characterized by a normal response of the erythroid progenitors to elevated levels of cytokines. Secondary polycythemia is usually the result of a physiologic response to chronic hypoxia (i.e., cyanotic CHD or sleep apnea), autonomous erythropoietin production (tumor secretion), or exogenous erythropoietin administration. Rarely, secondary polycythemia can be due to Hb variants with an altered affinity for oxygen or genetic mutations that result in disordered hypoxic sensing.
As previously mentioned no studies have defined the optimal hematocrit in patients with cyanotic CHD. Patients with cyanotic heart disease can develop a significant increase in the hematocrit leading to hyperviscosity and its associated morbidities including thrombotic complications and end-organ damage. Polycythemia treatment is clearly indicated in symptomatic patients (headache, visual disturbance, bone pain, fatigue, or thrombosis) and most commonly includes phlebotomy with a goal to reduce the hematocrit to <65%. Chronic phlebotomy over time can lead to iron deficiency which may increase a patient’s risk for thrombosis secondary to increased viscosity from the rigidity of abnormal red cells produced during iron restricted hematopoiesis (53,54,55). Although a small study in 39 adult patients with cyanotic heart disease found that blood viscosity did not differ when comparing those patients who did or did not have iron deficiency (56). Alternatively the use of hydroxyurea, which lowers red cell production through marrow suppression, has been reported and appears to be efficacious and safe although no prospective trial confirming these findings has been completed (57,58).
Disorders of White Blood Cells
Two genetic disorders, Barth syndrome and 22q11.2 deletion syndrome, may have both heart disease and associated white cell disorders. Barth syndrome is an X-linked recessive mitochondrial disorder that is characterized by a dilated cardiomyopathy (or sometimes hypertrophic) with endocardial fibroelastosis, skeletal myopathy, growth retardation, neutropenia, and organic aciduria (59,60). It is secondary to mutations in the TAZ gene that is responsible for remodeling the mitochondrial phospholipid: cardiolipin (61). The etiology of neutropenia in Barth syndrome is unclear at this time. Treatment can include granulocyte colony-stimulating factor to prevent severe neutropenia.
Chromosome 22q11.2 deletion syndrome encompasses a heterogeneous group of disorders. The most common phenotypic features include conotruncal cardiac malformations, immunologic dysfunction, developmental delay, and palate deformities (62). Immunologic dysfunction is the result of thymic aplasia or hypoplasia resulting in a variable T-cell deficiency with a resultant increase in infections and autoimmune disease (63). Decreased regulatory T cells may play a role in the increased incidence of autoimmune disorders (63,64). Autoimmune disease can include acquired cytopenias (65,66).
Disorders of Hemostasis Associated with Bleeding
Hemostatic derangements complicated with bleeding are very common in pediatric patients with heart disease. The holy grail of hemostatic testing would be one test that is rapidly available and adequately reports on all components of hemostasis. Unfortunately, this test does not exist, leaving clinicians with a series of coagulation tests that reflect various aspects of hemostasis.
Hemostatic testing: The PT and PTT serve as screening tests for procoagulant factor deficiencies. The PT and PTT do not provide a global picture of hemostasis. Proper specimen handling is imperative for coagulation assays. Heparin contamination of a specimen is the most common reason for a prolonged PTT in a hospitalized patient. Heparinase can be used to remove heparin from a specimen. Coagulation assays are also sensitive to the plasma-to-citrate ratio in the specimen tube. Underfilling of the specimen tube will falsely prolong the coagulation assays. Patients with significant polycythemia (hematocrit >55%) can also have an altered plasma-to-citrate ratio and will have falsely prolonged coagulation assays. A special specimen tube with a decreased amount of citrate should be used for patients with significant polycythemia. This is of particular concern when using the prothrombin time/international normalized ratio (PT/INR) to adjust warfarin dosing in cyanotic/polycythemic patients. Factor deficiencies that can cause an isolated prolonged PTT include either a deficiency in factor VIII, IX, XI, XII, prekallikrein, or high–molecular-weight kininogen. An isolated prolonged PT is secondary to factor VII deficiency. Prolongation in both the PT and PTT can be seen in the setting of factor II, V, X, or fibrinogen deficiency.
For monitoring unfractionated heparin (uFH) therapy, the PTT is commonly used as a surrogate marker of heparin activity. For therapeutic heparin, the PTT is titrated to 1.5 to 3 times control. There are limitations to the PTT because it is not a direct measure of heparin and it is affected by variables other than heparin including an elevated factor VIII, fibrinogen, or a Lupus anticoagulant. For example, when the FVIII level is >250%, the PTT is significantly shortened and will no longer reflect the heparin effect. (Factor VIII is an acute phase reactant and can be elevated in inflammatory states.) For a direct measure of heparin activity against factor X, the uFH anti-Xa level can be used with a recommended therapeutic range of 0.35 to 0.7 U/mL (note this is different from the LMWH anti–Xa-recommended therapeutic range of 0.5 to 1 U/mL).
The activated clotting time (ACT) is used to monitor higher heparin doses given to patients undergoing cardiac surgery with cardiopulmonary bypass (CPB), cardiac catheterization, or extracorporeal membrane oxygenation (ECMO). The ACT is a whole blood clotting time that is simple to perform with a rapid turnaround time. It is more sensitive to a wider range of heparin doses than the PTT. The limitations to this assay are that it is imprecise, proper specimen handing and timing are imperative, it is affected by the same variables that can prolong the PTT assay, and it is affected by the platelet count.
Fibrinogen is the final procoagulant protein in the coagulation pathway. It is converted by thrombin into fibrin. Fibrin is then cross linked by factor XIII to make a stable clot. In the setting of a bleeding patient, it is imperative that the fibrinogen level is maximized to ensure adequate hemostasis. The thrombin time measures the conversion of fibrinogen to fibrin and is affected by quantitative or qualitative abnormalities of fibrinogen, the presence of thrombin inhibitors, and fibrinogen degradation products. Heparin contamination of the specimen can prolong the thrombin time.
For an assessment of primary hemostasis, a platelet count indicates the number of platelets that are available but does not provide any information regarding platelet function. At this time, there is limited availability to rapidly assess platelet function. The bleeding time assesses platelet and capillary function but this technique is patient- and operator-dependent and has fallen out of favor (67). It is insensitive to mild platelet defects and does not consistently predict a bleeding tendency (68). The platelet function analyzer (PFA-100) is an in vitro screen for platelet function (68). The PFA-100 is prolonged in the setting of anemia or thrombocytopenia and is insensitive to mild platelet aggregation defects; it has also fallen out of favor as a screening test.
Thromboelastography (TEG) is a whole blood point-of-care assay that provides a real-time graph of clot formation and stability. Values measured include R (time necessary for initial clot formation), K (clot kinetics), α (rate of clot formation), maximum amplitude (maximum strength of the clot), and A-60 to maximum amplitude ratio (fibrinolysis). TEG has been utilized in pediatric CPB and in patients with LV assist devices (69,70,71).
Platelet mapping assay is a modification of the standard TEG and is used clinically to assess the effectiveness of antiplatelet medications. This assay utilizes whole blood that is heparinized to suppress thrombin generation, and then known platelet agonists are added to the sample to assess platelet activation. This assay is currently utilized most commonly in pediatric patients with an LV assist device to monitor antiplatelet therapy.
Acquired von Willebrand Disease
There is an increasing awareness of acquired von Willebrand disease (vWD) in patients with cardiovascular disorders. vWF is a procoagulant protein that has two important roles in hemostasis: it facilitates the binding of platelets to the site of vascular injury and it is the carrier protein for the coagulant protein factor VIII. vWF is initially secreted as a large multimeric protein that is further proteolyzed by ADAMTS-13 into variable sizes of multimers. The distribution in the size of these multimers is imperative for hemostatic balance. For example, a bleeding tendency is seen with decreased vWF multimer size (type 2a or 2b vWD) and a prothrombotic tendency is found when there is a persistence of ultra-large vWF multimers (congenital or acquired thrombotic thrombocytopenic purpura).
Most of the data on acquired vWD have focused on patients with aortic stenosis and with LV assist devices (72,73). There have been reports in neonates with patent ductus arteriosus, and in patients with endocarditis, dysfunctional valve prosthesis, septal defects, and mitral valve prolapse (74,75). The pathophysiology of acquired vWD in cardiovascular disorders is likely multifactorial but is thought to be secondary to lesions that result in increased shear stress. Increased shear stress results in the mechanical destruction of the larger vWF multimers and causes platelet activation that leads to the absorption of larger vWF multimers. Clinically, these patients will experience an increase in mucocutaneous bleeding and they could be at increased risk of hemorrhage with surgical procedures.
Laboratory findings in patients with cardiovascular disorders and acquired vWD reveal an abnormal multimer vWF distribution with a specific loss of the high–molecular-weight multimers. Commonly, the VW antigen and activity are normal to increased. Routine coagulation testing PT/INR, PTT, and platelet count will be normal. Definitive treatment of acquired vWD would be to correct the underlying cardiac abnormality but this may not always be plausible (76). Other treatment modalities would include desmopressin (DDAVP) that facilitates the release of endogenous stores of vWF, although the response rate is low (10%) in this setting (75). An alternative therapy consists of the use of plasma-derived vWF replacement product (75,77). Treatment is only necessary to treat active bleeding that does not resolve with local measures or prior to a significant hemostatic challenge to prevent excessive bleeding.
Platelet Disorders
Platelet disorders are either inherited or acquired. Congenital platelet disorders are very rare in the general population. Bernard–Soulier syndrome, a congenital platelet disorder, should be considered when caring for patients with 22q11.2 deletion syndrome. Bernard–Soulier syndrome is an autosomal recessive platelet disorder. The mutation is in the platelet GP 1b/IX complex that binds platelets to vWF. Children with 22q11.2 deletion syndrome can be heterozygotes for the Bernard–Soulier mutation given the proximity of the gene encoding the GP 1b/IX complex (78). Heterozygotes for this condition are clinically normal but may have mild thrombocytopenia and minor platelet function abnormalities (78). Patient with 22q11.2 deletion syndromes also have an increased prevalence of immune thrombocytopenia (ITP) (79,80).
In patients with cyanotic CHD, there is an inverse relationship between the platelet count and the degree of cyanosis. The lower the systemic arterial oxygen saturation, the higher will be the compensatory polycythemia. These physiologic changes are associated with a lower platelet count (81,82). Typically, the thrombocytopenia is not severe and the platelet count will be >50,000/μL. The thrombocytopenia can improve with phlebotomy especially when the hematocrit is >65% (83). The etiology of thrombocytopenia in cyanotic CHD with polycythemia is unclear at this time. Lill and Perloff hypothesize that right-to-left cardiac shunts bypass the delivery of whole megakaryocytes into the lungs thus reducing platelet formation through fragmentation in the pulmonary bed (81).
Drug exposure is a common reason for acquired platelet dysfunction or thrombocytopenia. Antiplatelet medications are commonly used in patients with heart disease to prevent thrombotic complications; these are discussed below. Numerous drugs have been implicated in causing thrombocytopenia. The more commonly used drugs include antibiotics (penicillins, or sulfa-containing antibiotics), antiepileptics (phenytoin, valproate, carbamezepine), H2 agonists (cimetidine or ranitidine), thiazide diuretics, and furosemide. In general, thrombocytopenia will resolve with removal of the offending drug. A special circumstance of drug-induced thrombocytopenia is heparin-induced thrombocytopenia (HIT).
Heparin-Induced Thrombocytopenia (HIT)
HIT is the result of antibodies that are formed against the heparin–PF4 complex that exists on platelets (84). It is proposed that the binding of antibodies to this platelet complex results in increased platelet reactivity and thus a prothrombotic state (84). This disorder is characterized by thrombocytopenia and resultant arterial and/or venous thrombosis that can be catastrophic. In adults, the prevalence of HIT is estimated to be 1% to 5% of patients exposed to heparin and untreated HIT has a mortality of 20% to 30% (85). The incidence of HIT in the pediatric population is not clearly defined but appears to be less than adults with a reported range of 0% to 2.3% (86,87,88,89). Higher-risk pediatric groups include patients undergoing CPB (90).
The diagnosis of HIT is based on clinical criteria. Multiple scoring systems have been developed and validated in adults but diagnostic criteria have yet to be established in the pediatric population (91,92,93). Classically, HIT is characterized by symptoms appearing 5 to 10 days post–heparin exposure with a 50% fall in the platelet count (rarely is the platelet count <50,000/μL) and there can be venous or arterial thrombosis. Laboratory testing can be supportive in making the diagnosis and includes ELISA for the heparin-P4 antibodies and the serotonin-releasing assay. The ELISA is readily available with a fast turnaround time. This test is highly sensitive but has a significant false-positive rate. The ELISA is most helpful in ruling out the diagnosis with a negative assay. The serotonin-releasing assay is highly specific and sensitive; it measures platelet reactivity in the presence of the patient’s plasma. Unfortunately, it is only performed in a few highly specialized laboratories so it is not readily available for most clinicians.
The treatment of HIT includes the removal of all heparin from the patient including avoidance of LMWH. Anticoagulation should be initiated with a nonheparin anticoagulant such as a direct thrombin inhibitor (i.e., bivalirudin or argatroban). In this setting, warfarin should never be initiated by itself due to an increased risk of skin necrosis and further thrombotic events. Warfarin can be initiated once the platelet count has normalized and overlapped with the nonheparin anticoagulant until the INR is therapeutic.
Additional causes of acquired thrombocytopenia in pediatric patients with heart disease include sequestration and consumptive causes. The spleen normally contains 30% of the platelet mass, and in the setting of an enlarged spleen, it can trap a larger portion of
platelets with resultant thrombocytopenia. In patients with heart disease, specific consumptive causes of thrombocytopenia include CPB and artificial heart valves or grafts. Other causes of consumption not necessarily specific to patients with heart disease include the use of ECMO, the presence of disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, hemophagocytic lymphocytic histiocytosis, HIV, or immune destruction of platelets.
platelets with resultant thrombocytopenia. In patients with heart disease, specific consumptive causes of thrombocytopenia include CPB and artificial heart valves or grafts. Other causes of consumption not necessarily specific to patients with heart disease include the use of ECMO, the presence of disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, hemophagocytic lymphocytic histiocytosis, HIV, or immune destruction of platelets.
Immune Thrombocytopenia
ITP is an immune-mediated destruction of platelets generated by the presence of IgG autoantibodies. These antibodies coat the surface of the platelets and these platelets are cleared through the Fcγ receptors in the spleen. In childhood, ITP is considered a benign self-limited disorder; 80% of patients spontaneously resolve within 6 months with only a small percentage (<20%) developing chronic ITP. Treatment can include observation combined with thrombocytopenic precautions or the use of immune-modulating therapies like immune globulin or steroids. In the neonate, immune destruction of platelets can be secondary to a mismatch between the fetal and maternal platelets, termed neonatal alloimmune thrombocytopenia. The mother develops antibodies against antigens on the fetal platelet surface that were inherited from the father. These antibodies cross in the placenta, coat the fetal platelets, and result in significant thrombocytopenia. Thrombocytopenia can also be the result of decreased production that can be found in aplastic anemia, myelodysplastic syndrome, infiltrative marrow processes (i.e., leukemia), or nutritional deficiency states.
Thrombocytosis
Thrombocytosis is defined as an increase in the platelet count that is two standard deviations above the mean value for age. Thrombocytosis can be primary or secondary. Primary causes include myeloproliferative disorders that are either inherited (essential thrombocythemia) or acquired (polycythemia vera, leukemia, or myelodysplastic syndrome). Secondary causes are the result of underlying inflammation (infection, Kawasaki disease, rheumatologic disorders, or inflammatory bowel disease), hematologic disorders (hemolytic anemia or iron deficiency), drugs (vinca alkaloids or corticosteroids), or decreased splenic pooling in the setting of asplenia.
CPB, ECMO, and Ventricular Assist Devices
Postoperative Bleeding after CPB
Postoperative bleeding is a common complication in pediatric patients undergoing CPB and is associated with increased morbidity and mortality (94,95). CPB is a nonphysiologic state and the passage of blood through the artificial circuit results in the activation of the coagulation, fibrinolytic, and inflammatory systems. The coagulation system is activated through the contact pathway and ultimately results in the formation of fibrin. With fibrin deposition within the CPB circuit, there is platelet adherence and activation. Prematurely activated platelets are no longer available for hemostasis.
Post-CPB bleeding is more common in pediatric patients than in adults. Clinical factors associated with postoperative bleeding include younger age (<1 year), lower weight (<8 kg), cyanosis, and prolonged circuit and deep hypothermia times (96,97,98). One major contributing factor is the degree of hemodilution that occurs in pediatric patients with the institution of cardiac bypass. This is secondary to the large discrepancy between a pediatric patient’s blood volume and the circuit volume. An adult patient experiences approximately 25% hemodilution with the institution of bypass, while a pediatric patient can experience up to 60% of hemodilution (99). This dilution is further exacerbated by developmental hemostasis in neonates who, at baseline, already have lower procoagulant and anticoagulant proteins (4).
To prevent postoperative CPB bleeding, multiple approaches have been utilized including adequate anticoagulation during CPB, modified ultrafiltration, adequate reversal of anticoagulation post-CPB, transfusion of blood components, and antifibrinolytics (99). High-dose heparin is used during CPB to prevent thrombosis of the circuit. Heparin exerts its anticoagulant effect through the binding of antithrombin. Neonates developmentally have lower antithrombin levels that can contribute to a variable heparin effect. There are also significant issues with monitoring heparin therapy during CPB in pediatric patients. It has been shown in children that the ACT values do not correlate with heparin levels. Specifically, the ACT overestimates the heparin effect because it is prolonged by other variables including hemodilution and hypothermia (99,100,101). Adequate reversal of heparin with protamine is dependent on knowing the correct heparin concentration. Too little protamine means heparin is still circulating or excessive unbound protamine has anticoagulant properties (102,103). Hyperfibrinolysis is also an important mechanism that leads to post-CPB bleeding (104,105). All three antifibrinolytic therapies (aprotinin, tranexamic acid, and aminocaproic acid) have been shown to decrease post-CPB hemorrhage (106). Aprotinin was removed from the US market in 2008 because of its association with renal failure and death in adult recipients (106,107). Despite the use of these therapies, hemorrhage following CPB remains a frequent occurrence.
Management of Postoperative Bleeding
Although some bleeding from indwelling mediastinal drains is expected after cardiac surgery, the rate of bleeding should decrease as each postoperative hour goes by. Excessive bleeding is a clinical concern that warrants immediate attention and constant vigilance. In the immediate postoperative period, bleeding of <5 mL/kg/h is often associated with minor abnormalities in coagulation status. Red cell transfusion may be necessary to correct a postoperative anemia but blood component administration is rarely necessary. Bleeding 5 to 10 mL/kg/h should prompt notification of the cardiothoracic surgeon and continued evaluation of the patient at the bedside. Any abnormalities in coagulation parameters should be corrected. Blood loss must be balanced by RBC transfusion. The patient must be closely monitored for persistence or an increase in the rate of bleeding that may signal the presence of a surgical bleeding site or may be the result of loss of coagulation factors secondary to the ongoing hemorrhage. Bleeding of >10 mL/kg/h that persists or increases will likely result in hemodynamic compromise if not abated. The cardiothoracic surgeon should decide whether reexploration is needed to exclude a bleeding site or to remove thrombus that may be perpetuating further bleeding. The cardiac intensivist must exclude concomitant hemopericardium and/or hemothorax (often herald by tachycardia, hypotension, desaturation, and/or tamponade physiology) while keeping up with the blood loss (RBC transfusion) and coagulopathy (blood component replacement). Even after the decision has been made to reoperate, the bedside medical team must continue to be vigilant with RBC, volume, and blood component replacement as indicated by hemodynamic parameters and laboratory data.
Recombinant factor VIIa (rFVIIa, eptacog alfa [activated], NovoSeven, Novo Nordisk Pharmaceuticals Inc., Princeton, NJ) is currently utilized as an off-label hemostatic agent in the management of postoperative CPB bleeding. It is an analog of the naturally occurring procoagulant factor VII and induces hemostasis by binding to exposed tissue factor at the site of endothelial injury and stimulating thrombin formation (108,109). Current studies indicate
that children and adults with cardiovascular disease account for a large proportion of off-label rFVIIa use (110,111).
that children and adults with cardiovascular disease account for a large proportion of off-label rFVIIa use (110,111).
There are concerns regarding the thrombotic potential of rFVIIa and this concern is further heightened in pediatric patients with CHD because of a known increased incidence of thrombosis (112). Current evidence to support off-label use of rFVIIa in pediatric CPB is limited. Only one pediatric randomized clinical trial of rFVIIa in CPB has been conducted and it was found that there was no benefit to the prophylactic use of rFVIIa among 82 infants undergoing surgery for correction of CHD (113). The primary end point, time to chest closure, was actually prolonged in the treatment group. No difference was noted in the secondary end points of surgical blood loss or use of blood products (113). A recent systematic review regarding the off-label use of rFVIIa in pediatric cardiac surgery reviewed 29 studies that included a total of 169 patients (114). The authors concluded that there is no supporting evidence to use rFVIIa prior to cardiac surgery to prevent bleeding. Randomized clinical trials should be performed that utilize appropriate clinical outcomes to address the use of rFVIIa to treat bleeding after cardiac surgery. Therapeutic options for postoperative bleeding are listed in Table 75.2.
ECMO and Ventricular Assist Devices
Patients on ECMO or who have ventricular assist devices (VADs) are at increased risk for both bleeding and thrombotic complications. Similar to CPB, both processes have the passage of blood through an artificial circuit resulting in activation of coagulation. Anticoagulation remains the mainstay to prevent thrombotic outcomes. In ECMO, uFH is commonly used for anticoagulation. In patients with a VAD, one type of anticoagulation (uFH, LMWH, or warfarin) is commonly combined with antiplatelet therapy although anticoagulation management will vary by specific device manufacturer recommendations.
TABLE 75.2 Therapeutic Options for Postoperative Bleeding | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
Thrombosis
Thrombosis in Children and Adolescents with Congenital and Acquired Heart Disease
Thrombosis has long been recognized as a clinical problem to those caring for adolescents and children with congenital and acquired heart disease. Much work has focused on the diagnosis, treatment, and prevention of thrombosis in Kawasaki disease (115,116,117,118,119,120,121,122,123,124,125) and to a lesser extent on the thrombotic complications associated with cardiac catheterization (126,127,128,129,130,131,132,133,134,135,136) and cardiomyopathies (137,138,139,140,141).
The prevention and management of thromboses related to prosthetic valves (142), arrhythmias (143,144), and pulmonary hypertension (145,146,147,148,149,150) in children has largely been extrapolated from the adult literature. For the past decade, the single-ventricle population has been identified as a particularly high-risk population for thrombosis and their potentially devastating sequelae (151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


