Heart Muscle Diseases
Classification
Heart muscle diseases include a diverse range of cardiomyopathies and myocardites. Cardiomyopathies have been previously classified as diseases of unknown cause and were therefore distinct from more specific causes of heart muscle disease such as ischaemia, hypertension, and valvular heart disease. However, a better understanding of their aetiology and pathophysiology has led to this distinction becoming obsolete.
Definition
Cardiomyopathies can be defined as a heterogeneous group of diseases of the myocardium, characterized by mechanical and/or electrical dysfunction that typically (though not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that are frequently genetic. Cardiomyopathies are either confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability (Fig. 8.1).1
Cardiomyopathies can be classified according to the predominant organ involvement. Primary cardiomyopathies are those confined to the heart muscle and can be further subdivided into genetic and acquired forms. Secondary cardiomyopathies occur as part of a variety of generalized systemic diseases, often with multi-organ involvement, which also demonstrate myocardial pathology.
Primary cardiomyopathies
Primary cardiomyopathies are predominantly confined to the heart only. They can be further classified into genetic and acquired groups, though overlap exists with certain forms of cardiomyopathy, in particular dilated cardiomyopathy.
Secondary cardiomyopathies
Secondary cardiomyopathies are typically part of generalized multisystem diseases. Cardiac manifestations may be an isolated feature, though multiple organ involvement is more common. Conversely, if no evidence of myocardial disease is found, regular surveillance for cardiac involvement with history, examination and non-invasive investigations is important. Fig. 8.2 lists examples of conditions associated with secondary cardiomyopathy but is not exhaustive.
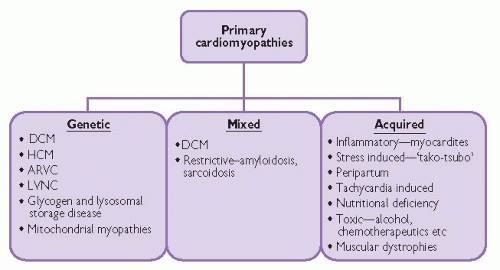 |
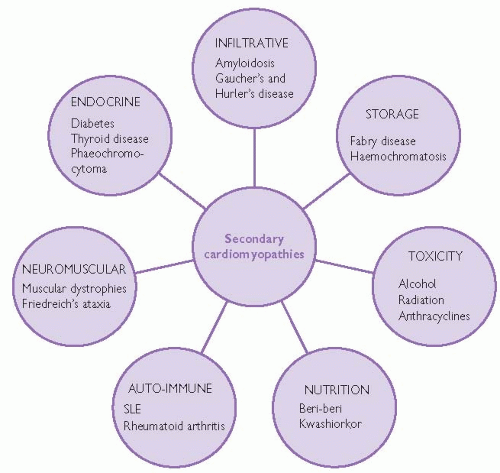 Fig. 8.2 Secondary cardiomyopathies can be a feature of many generalized systemic diseases. |
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is characterized by cardiac chamber enlargement and impaired systolic dysfunction, although diastolic dysfunction is almost always also present. The prevalence is 5-8 per 100 000 and it is 3 times more frequent in black and male than white and female individuals.
Symptoms
Clinical presentation may be abrupt, with acute pulmonary oedema, systemic or pulmonary emboli, or even sudden death, but more often patients present with progressive symptoms of congestive cardiac failure (CCF) including exertional dyspnoea, orthopnoea, paroxysmal nocturnal dyspnoea, and fatigue. Right upper quadrant discomfort, nausea, and anorexia may relate to hepatic congestion. Syncope is an ominous symptom and should be regarded to represent a potentially fatal arrhythmia unless subsequent investigation indicates otherwise.
Diagnosis
Diagnosis is established by physical examination, electrocardiography, chest X-ray (CXR), and echocardiography.
A raised jugular venous pressure (JVP), displaced cardiac apex, and added heart sounds are sensitive clinical markers of heart failure. Crackles in the lungs and swollen ankles are relatively non-specific, especially in the octogenerain population.
Electrocadiography (ECG) may show evidence of sinus tachycardia, atrial fibrillation (AF), or frequent ventricular extra-systoles. Voltage criteria for cardiac chamber enlargement, bundle branch block, nonspecific T-wave changes, and poor R-wave progression in the anterior chest leads are common.
CXR may show an enlarged cardiac size and pulmonary oedema (upper lobe venous diversion, interstitial oedema, pleural effusions and Kerley B lines).
Echocardiography (ECHO) is currently regarded to be the gold standard investigation for diagnosing left ventricular (LV) dysfunction. Bi-atrial and biventricular enlargement are common, and patients with chronic LV volume overload may also exhibit mild left ventricular hypertrophy (LVH). Indices of systolic (and diastolic function) are impaired. The atrioventricular (AV) valves are commonly incompetent, due to annular dilatation. ECHO may reveal complications of dilated cardiomyopathy such as intramural thrombus, and is invaluable in the identification of alternative causes of heart failure, such as hypertensive heart disease, previous myocardial infarction (MI), valvular heart disease, pericardial disease, and intracardiac shunts.
Exercise testing with or without measurement of maximum ventilatory oxygen consumption, is useful to assess functional capacity and prognosis.
Ambulatory ECG monitoring is essential to identify the presence of paroxysmal AF and non-sustained ventricular tachycardia (VT), which have important therapeutic and prognostic implications respectively.
There is rarely any indication for cardiac catheterization in patients with dilated cardiomyopathy, with the exception of excluding coronary artery disease.
Aetiology of dilated cardiomyopathy
Although originally considered to be idiopathic, experimental and clinical data now suggest that genetic, viral, and autoimmune factors play a role in its pathophysiology. Several genetic mutations have been identified as the causative problem in familial DCM, while certain viruses have been shown to cause sporadic cases of DCM. The following list is not exhaustive and there is some overlap with specific cardiomyopathies.
Inherited (may account for >25% of cases)
Myocarditis (infective, autoimmune, toxic)
Metabolic (haemochromatosis, thyrotoxicosis)
Nutritional (vitamin deficiencies—thiamine (beri-beri))
Persistent tachycardia (tachymyopathy)
DCM is essentially a diagnosis of exclusion, and potentially reversible causes including coronary artery disease, valvular heart disease, and adult congenital heart disease should be sought. Careful attention should also be paid to dietary history and alcohol consumption, as some reversibility is possible with modification of these factors.
Additional investigations for DCM—the ‘cardiomyopathy screen’
Renal function
Liver function tests (LFTs)
Serum ferritin, iron, transferrin, B12, and folate
Thyroid function
Viral serology
Infective screen (HIV, hepatitis C, enteroviruses)
Autoantibodies
Dilated cardiomyopathy: treatment
Management of DCM focuses on relieving symptoms and improving prognosis and quality of life. Pulmonary and peripheral congestion are effectively treated with diuretics. Prognostically important pharmaceutical therapies inhibit the maladaptive neurohormonal process involving the sympathetic system and rein-angiotensin-aldosterone axis. The acute management of heart failure is discussed in  Chapter 7, p.367.
Chapter 7, p.367.
Chapter 7, p.367.Diuretics
Loop diuretics, are useful in relieving symptoms caused by pulmonary and peripheral congestion. Careful monitoring of electrolytes is important, as intravascular depletion may cause urea to rise, and hypokalaemia is common. Hypokalaemia may be counteracted by the co-administration of a potassium-sparing diuretic such as amiloride or spironolactone. Spironolactone is an aldosterone antagonist. The recent RALES (Randomized Aldactone Evaluation Study) trial (see  p. 106) demonstrated that the use of low-dose spironolactone can reduce mortality in patients with severe CCF.
p. 106) demonstrated that the use of low-dose spironolactone can reduce mortality in patients with severe CCF.
p. 106) demonstrated that the use of low-dose spironolactone can reduce mortality in patients with severe CCF.Angiotensin-converting enzyme inhibitors
There is overwhelming scientific evidence that angiotensin-converting enzyme inhibitors (ACE-Is) improve symptoms and outcomes in patients with heart failure, irrespective of symptoms. First-dose hypotension used to be a concern but this is rarely observed with newer ACE-Is and usually only in patients who are relatively intravascularly depleted due to the concomitant use of high-dose diuretics. Side-effects include a dry cough, which may affect up to 20% of patients and is due to increased levels of bradykinin. Angioedema is a rare but potentially life-threatening complication of ACE-Is. Angiotensin receptor 1 antagonists (ARBs) can be used as an alternative in patients who experience a dry cough.
Contra-indications to ACE-I include established bilateral renovascular disease, severe aortic stenosis, pregnancy, and a baseline potassium >6 mmol/L. ACE-Is should be prescribed with caution in individuals with a resting systolic blood pressure <90 mm Hg or basline creatinine >200 µmol/L.
Beta-blockers
Beta-blockers also provide symptomatic and prognostic benefit and are recommended in all patients with dilated cardiomyopathy unless there are specific contraindications. Beta-blockers are effective through several mechanisms, which include a reduction of myocardial oxygen consumption, enhanced LV filling, inhibition of the apoptotic effects of catecholamines on cardiac myocytes, treatment of cardiac arrhythmias, and upregulation of beta-1 receptors. Given the potentially negative inotropic effects, the drugs are initiated in low doses, which are titrated up gradually. They should not be started in patients with overt heart failure.
Aldosterone antagonists
Alodosterone antagonists improve symptoms and prognosis and are recommended in patients who continue to remain in NYHA (New York Heart Association) class III despite adequate doses of ACE-Is and betablockers. The most important complication is hyperkalaemia due to the concomitant use of ACE-I. Painful gynaecomastia is a recognized sideeffect, particularly in males taking digoxin and anti-androgens.
Antiarrhythmic agents
Antiarrhythmics agents have not been shown to reduce the incidence of sudden cardiac death (SCD) in patients with DCM. AF is a common arrhythmia in DCM and may be associated with symptoms of cardiac decompensation. Most individuals are rate controlled with β-blockers, although digoxin may be used as additive treatment in individuals who continue to exhibit rapid heart rates despite satisfactory doses of β-blocker. Maintenance of sinus rhythm is usually unsuccessful in the long term.
Anticoagulation
Patients with DCM are prone to thromboembolic complications and should be anticoagulated with warfarin regardless of underlying rhythm.
Device therapy
Cardiac resynchronization therapy (CRT) utilizes biventricular pacing to combat intraventricular and interventricular dys-synchony associated with ventricular conduction defects. CRT has been shown to be associated with significant improvements in functional capacity and rates of rehospitalization from heart failure ( see p. 404). CRT is generally reserved for individuals with wide QRS complexes (particularly left bundle branch block (LBBB)) who have an ejection fraction (EF) of <35% and are in NYHA class III despite optimal medical therapy.
see p. 404). CRT is generally reserved for individuals with wide QRS complexes (particularly left bundle branch block (LBBB)) who have an ejection fraction (EF) of <35% and are in NYHA class III despite optimal medical therapy.
Cardiac transplantation
Orthotopic cardiac transplantation using an allograft can be considered in patients who are severely symptomatic despite maximal medical therapy. The limited availability of donor organs still restricts its role. As a result, there is considerable interest in using organs from other species (xenografts). However, technical hurdles still remain before this can be considered a viable option. The artificial heart is another area that has received much interest and publicity, and clinical trials are soon to be conducted.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is characterized by unexplained LVH and has a prevalence of 1 in 500. HCM is a heterogeneous disorder in terms of clinical manifestation, cardiac morphology, and natural history. Whereas the vast majority of affected individuals have a relatively normal life span, HCM is most recognized for being the commonest cause of exercise-related SCD in young individuals under 35 years of age.
Genetics
HCM exhibits marked allelic and non-allelic heterogeneity, with multiple mutations in at least 12 genes encoding sarcomeric contractile protein. The majority of the mutations (>70%) are in the β-myosin heavy chain, troponin T and myosin-binding protein C genes (see Table 8.1).
Pathophysiology
The macroscopic hallmark of HCM is LVH, which usually affects the interventricular septum in an asymmetric fashion; however, almost any pattern of LVH is possible, including concentric LVH as seen in individuals with hypertension, and hypertrophy localized to only 1 or 2 myocardial segments. The magnitude of LVH is also variable and can range from very severe LVH (>30 mm) to very mild LVH (13-15 mm). There are also recognized familial cases where the only manifestation of the disorder is an abnormal ECG. Individuals with HCM often exhibit elongated mitral valve leaflets. Histologically, there is evidence of myocyte disarray, myocardial scarring, and abnormal intramyocardial arterioles. Together, these abnormalities manifest as:
hyperdynamic systolic function, which may be associated with
left ventricular outflow tract (LVOT) obstruction due to systolic anterior motion (SAM) of the anterior mitral valve leaflet (present in 725% of patients at rest but up to 70% during exercise)
impaired myocardial relaxation and raised filling pressures
myocardial ischaemia, and
LVOT obstruction occurs as a consequence of forward motion of the anterior mitral leaflet towards the hypertrophied proximal interventricular septum in systole. There are two main postulated mechanisms, which include (1) anterior displacement of the papillary muscles and (2) the so-called Venturi effect, created by rapid ejection of blood across a narrow LVOT, which ‘sucks’ the anterior mitral valve leaflet against the septum. See also Table 8.2.
Symptoms
Patients with HCM are often asymptomatic and often identified incidentally during routine medical examination, an abnormality on the ECG, or through family screening following the diagnosis in a first-degree relative.
Recognized symptoms may include:
fatigue and breathlessness due to impaired diastolic filling and decreased cardiac output. Atrial transport is very important for maintaining cardiac output, and symptoms typically deteriorate with AF
chest pain (angina) may result from increased cardiac work secondary to the LVH, a relative blood supply-demand mismatch, and narrowing of intramural arterioles. High diastolic pressures increase the diastolic wall stress and impair diastolic coronary blood flow
palpitations, pre-syncope, and syncope may occur due to atrial and ventricular arrhythmias or mechanical obstruction to cardiac output in those patients with left ventricular outflow pressure gradients
sudden death may be the initial presentation
approximately 5% of patients may eventually develop progressive left ventricular dilatation and failure, due to ongoing myocardial fibrosis and chronic small-vessel ischaemia.
Physical examination
Look for LVH (a forceful apical impulse and an S4 heart sound). Outflow tract obstruction is evident by a double apical impulse and ejection systolic murmur beginning in mid-systole, which may be augmented by provocation by manoeuvres such as Valsalva or squatting. A pansystolic murmur due to mitral regurgitation resulting from the SAM of the mitral valve may also be present.
Table 8.2 Dynamic manoeuvres to assess LVOT obstruction | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Hypertrophic cardiomyopathy: investigations
ECG
ECG is abnormal in >95% of cases. Isolated Sokolow-Lyon criterion for LVH is identified in only 2% of cases. Features include:
ST- and T-wave abnormalities
LVH with Romhilt-Estes points’ score >5
pathological Q waves in inferior and lateral leads (septal hypertrophy)
deep T-wave inversions (particularly in anterior and inferior leads in the apical form of HCM)
pre-excitation and Wolff-Parkinson-White (WPW) syndrome
ventricular ectopics
atrial fibrillation.
Echocardiography
Echocardiography continues to remain the gold standard investigation due to its widespread availability. The investigation is usually for detection of the presence, magnitude, and distribution of LVH, as well as identifying patients with basal LVOT obstruction. Recognized echocardiographic features include:
asymmetric septal hypertrophy (ASH): grossly thickened septum compared with posterior LV wall, with reduced septal motion; however, almost any pattern of LVH is possible
small LV cavity
SAM: systolic anterior movement of the mitral valve apparatus
mid-systolic aortic valve closure or fluttering of the aortic valve leaflet tips
impaired diastolic function and left atrial enlargement.
Cardiac magnetic resonance imaging (MRI)
Other investigations
Other investigations may help in risk stratification, although no single investigation accurately predicts those at risk of SCD, and negative tests do not entirely exclude the risk of SCD:
ambulatory ECG monitoring—AF and ventricular tachycardia arrhythmias. Non-sustained VT is a risk factor for SCD
exercise testing—functional capacity and blood pressure response to exercise. A flat blood pressure response during exercise (failure of the systolic blood pressure to rise by 25 mmHg from rest to peak exercise, or a paradoxical drop in systolic blood pressure during exercise) is a recognized risk marker for SCD
there is no role for cardiac catheterization in the diagnosis of HCM, although coronary angiography may be performed in adult patients with angina to exclude co-existent coronary artery disease (CAD)
there is no role for electrophysiology studies in the diagnosis of HCM. Ventricular stimulation studies have a poor predictive accuracy for the identification of high-risk patients; however, radiofrequency ablation of accessory pathways and atrial flutter circuits may be therapeutically important
genetic testing is being utilized increasingly; however, a genetic diagnosis is currently only possible in 60-70% of cases. Genetic testing is particularly useful for cascade screening of family members if a causative gene in the proband can be identified.
Hypertrophic cardiomyopathy: treatment
Approximately half of sudden deaths in HCM occur during or shortly after strenuous exercise, and therefore patients should be advised against competitive sports of a high-dynamic and high-intensity nature. The management of HCM includes (1) amelioration of symptoms including abolition of LVOT obstruction; (2) treatment of arrhythmias; (3) identification of individuals at risk of SCD, with a view to implantation of ICD; and (4) screening first-degree relatives for the disorder.
Treatment for symptoms
β-blockers
β-blockers are the mainstay of therapy for angina, dyspnoea, giddiness, and syncope. They reduce myocardial oxygen demand and improve diastolic filling, and are effective in the treatment of angina and exertional dyspnoea. Large doses may be required.
Calcium-channel antagonists
Calcium-channel antagonists (verapamil and diltiazem) are as effective as β-blockers and may be used in patients in whom β-blockers are not tolerated or are contraindicated.
Management of arrhythmias
Beta-blockers and amiodarone are the anti-arrhythmic agents of choice in the management of supraventricular arrhytyhmias. None of these agents have been shown to reduce the risk of SCD. AV re-entrant tachycardia and atrial flutter are usually amenable to radiofrequency ablation. Sustained VT is treated with amiodarone but such patients are candidates for ICD; the amiodarone reduces the frequency of non-sustained ventricular tachycardia (NSVT) and the need for recurrent anti-tachycardia pacing. Patients with AF should be anticoagulated with warfarin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


