(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Besides their central bioenergetic role in supplying ATP, heart mitochondria play an essential role in the regulatory and signaling events that occur in response to physiological stresses, including but not limited to heart failure (HF), myocardial ischemia and reperfusion injury (IRI), hypoxia, oxidative stress (OS), and hormonal and cytokine stimuli. Research on both intact cardiac and skeletal muscle tissue and cultured cardiomyocytes has just begun to probe the nature and the extent of mitochondrial involvement in interorganelle communication, hypertrophic growth, and cell death. In this chapter we will discuss mitochondria participation, at the molecular and biochemical levels, in the multiple and interrelated signaling pathways, gauging the effect that mitochondria have as a receiver, integrator, and transmitter of signals on cardiac phenotype.
Introduction
Mitochondria, a dynamic receiver and integrator of numerous translocated signaling proteins (including protein kinases and transcription factors), regulate Ca2+ fluxes and membrane phospholipids and transfer mitochondria-generated reactive oxygen species (ROS) and energy-related signaling. The contribution of mitochondria to cardiac function extends beyond their critical role as a bioenergetic supplier of ATP. The organelle plays an integral part in the regulatory and signaling events that occur in response to physiological stresses including but not limited to HF (see Chap. 18), myocardial ischemia reperfusion injury (IRI), hypoxia, OS, and hormonal and cytokine stimuli. Research on intact cardiac muscle tissue and in cultured cardiomyocytes has demonstrated the nature and the extent of mitochondrial role in interorganelle communication (see Chap. 6), hypertrophic growth, and cell death. Alongside their critical participation in cardiac and skeletal muscle apoptosis, mitochondria play a significant role in proliferative signaling pathways, in nutrient sensing, and in the responses of cells to metabolic changes and physiological stresses. In this chapter the participation of mitochondria in numerous and interrelated signaling pathways; the effect that these organelles have as receivers, integrators, and transmitters of signals on cardiac phenotype; and their potential impact on the treatment of cardiac diseases will be discussed.
Mitochondria Signaling
Without receptor molecules, mitochondria would be isolated and unaware of their environment. Signaling or communication pathways permit mitochondria to respond to the heart’s high-energy demands, cardiomyocyte growth, death, and physiological stimuli and stresses. To better understand mitochondrial signaling in the context of diverse and numerous intracellular events, we will describe some aspects of mitochondrial bioenergetics and biogenesis, as well as the organelle involvement in reactive oxygen species (ROS) generation and apoptosis. Thereafter, we will analyze the many roles that mitochondria play up as a receiver, integrator, and transmitter of signals and the interaction of signals from translocated proteins, such as protein kinases, receptors, and transcription factors; regulatory Ca2+ fluxes and membrane lipids; mitochondria-generated ROS; and energy-related communication with other intracellular compartments. New experimental approaches to probe mitochondrial signaling using metabolic inhibitors and genetic stresses such as mitochondrial DNA (mtDNA) depletion will also be outlined.
Mitochondrial Bioenergetics
Because mitochondrial bioenergetics has been extensibly discussed in Chap. 5, it suffices to point out here that energy production by the organelle is depending on both nuclear- and mtDNA-encoded genetic factors—which modulate normal mitochondrial function, including enzyme activity and cofactor availability—and on environmental factors such as substrate availability of sugars, fats, proteins, and oxygen. Several interacting bioenergetic pathways contribute to mitochondrial energy metabolism including pyruvate oxidation, the tricarboxylic acid (TCA) cycle, fatty acid β-oxidation (FAO), and the common final pathway of oxidative phosphorylation (OXPHOS), which generates most of the cellular ATP. OXPHOS is performed by complexes of proteins located at the mitochondrial inner membrane [1] including the electron transport chain (ETC)/respiratory complexes I–IV, the ATP synthase (complex V), and the adenine nucleotide translocator (ANT) Fig. 8.1. In order to be effectively utilized for bioenergetic production via mitochondrial FAO, fatty acids need to be transported into the cell and subsequently into the mitochondria, a process requiring several transport proteins including the carnitine shuttle (composed of carnitine acyltransferase and two carnitine palmitoyl transferases as well as carnitine). FAO and carbohydrate oxidation, via the TCA cycle, generates the majority of intramitochondrial NADH and FADH2, the direct source of electrons for the ETC.
Mitochondrial Biogenesis
Since mitochondrial biogenesis has been comprehensively discussed in Chap. 3, here it will suffice to mention that organelle biogenesis increases in specific cell types (e.g., cardiac and skeletal muscle) during cell hypertrophy and treatment with a variety of agents, such as thyroid hormone (TH), exercise, glucocorticoids, xenobiotics, and electrical stimulation [2–8]. While it is widely accepted that mtDNA copy number is highly regulated within a given cell or tissue type [9], the mechanisms that control specific mtDNA levels and overall mitochondrial copy number have not been established. On the other hand, it is well known that the nuclear genome encodes the full complement of proteins involved in mtDNA replication and transcription, the protein components of mitochondrial ribosomes, the multiple structural and transport proteins of the mitochondrial membranes, the remaining peptide subunits of the respiratory complexes (other than the 13 mtDNA-encoded peptide subunits), and the mitochondrial enzymes involved in mitochondrial lipid metabolism and the TCA cycle [10]. These nuclear-encoded proteins, synthesized on cytosolic ribosomes, are targeted to mitochondria and translocated to the organelle by an elaborated process, which involves signal peptide recognition, membrane receptors, proteases, and an arrangement of molecular chaperones [11] Fig. 8.1. Regulation of many of the nuclear-encoded OXPHOS proteins is mediated by changing gene expression, which is affected by an array of physiological (e.g., hypoxia) and developmental stimuli [12]. Tissue-specific isoforms for specific peptides (e.g., cardiac/skeletal muscle-specific isoforms exist for genes encoding cytochrome c oxidase subunits VIa, VIIa, and VIII) show entirely different patterns of gene expression in adult compared to the fetal stages of development [12, 13].
Signaling at the Mitochondria
Reactive Oxygen Species Generation and Signaling
Since a comprehensive discussion on the role of ROS in cell signaling, including their positive and negative effects, has been presented in Chap. 10, here it will suffice to note that ROS are critical by-products of mitochondrial bioenergetic activity and that side reactions of mitochondrial ETC enzymes with oxygen directly generate superoxide anion radical. Significantly, the primary sites for mitochondrial ROS generation, as a by-product of normal metabolism, are at complex I, II, and III of the respiratory chain, and either excessive or diminished electron flux at these sites can favor the auto-oxidation of flavins and quinones (including coenzyme Q) producing superoxide radicals [14].
Mitochondrial Receptors
Whereas a large number of receptors have been identified in different tissues and cell types, few well-characterized heart mitochondrial receptors have been hitherto detected. Interestingly, the thyroid hormone (TH) receptor Erb-Aa, which has been identified as an “orphan” receptor interacting with mtDNA during targeted hormonal stimulation (Fig. 8.1) [15], heretofore has not been found in cardiac tissue, notwithstanding the significant effect of TH in heart mitochondria. Moreover, a large number of nuclear transcription factors including p53, NF-κB, PPAR-α, RXR-a, and TR3 (an “orphan receptor” of the steroid-thyroid hormone-retinoid receptor superfamily of transcription factors), which have been characterized in many tissues/cell types as translocating to mitochondria, hitherto have not been found in heart mitochondria.
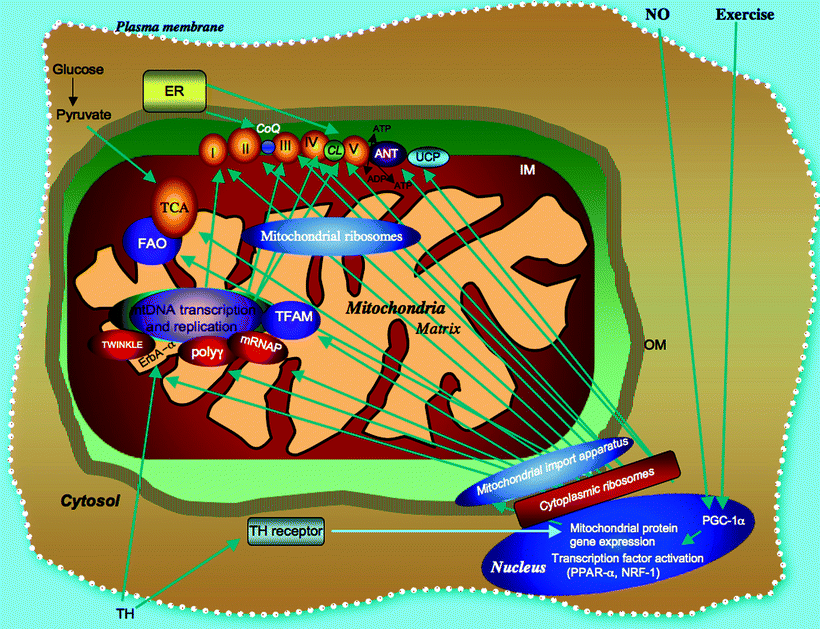
Fig. 8.1
Biogenesis and bioenergetic pathways of myocardial mitochondria are signal driven. The major mitochondrial bioenergetic pathways including the matrix-localized tricarboxylic acid (TCA) cycle, the inner membrane (IM)-associated fatty acid oxidation (FAO) pathway, and the respiratory complexes (I–V) with closely associated adenine nucleotide translocator (ANT) involved in ATP/ADP mitochondria transport, and uncoupling proteins (UCP) effecting proton transport are shown. Thyroid hormone (TH), exercise, and nitric oxide (NO) dependent activation of transcription factors in the nucleus [e.g., PPARγ coactivator 1 (PGC1), peroxisome proliferator-activated receptor (PPAR), and nuclear respiratory factor (NRF1)] and a subset of factors which activate mitochondrial DNA (mtDNA) transcription by interaction with D-loop sequences promoting mitochondrial biogenesis are also shown. All but 13 mitochondrial proteins are encoded in the nucleus, translated on cytoplasmic ribosomes, and incorporated through the outer membrane (OM) by a mitochondrial import apparatus. Other abbreviations: CoQ coenzyme Q, ER endoplasmic reticulum, ErbA-α thyroid hormone receptor-α, mRNAP mitochondrial RNA polymerase, poly γ DNA polymerase (subunit γ), TFAM transcription factor A of mitochondria, TH thyroid hormone, TWINKLE a hexameric DNA helicase
Although TNF-α can be induced in cardiomyocyte mitochondria [16], no specific mitochondrial receptor binding TNF-α or to various cytokines known to effect cardiac mitochondrial function have been found. Also, in contrast with other noncardiac tissues, characterization in heart mitochondria of anchoring proteins that bind and concentrate protein kinases has been rather limited. Further research on mitochondrial receptors and on the proteins associated with apoptosis and mitochondrial removal may increase our understanding of the role that these receptors and mitophagy play in normal physiological processes and in cardiac diseases [17]. For a comprehensive discussion on mitophagy, see Chap. 10.
Translocation of Proteins into Mitochondria
A unifying concept regarding mitochondrial signaling is the stimuli-generated translocation of specific cytosolic proteins into the organelle; the increasing list of translocated proteins includes many of the proapoptotic proteins (e.g., Bax, Bid) as well as protein kinases. Many of these kinases appear to target specific proteins on the outer mitochondria membrane, and others, imported as preproteins, are recognized by a small set of specific receptors (so-called translocases) on the mitochondrial outer membrane (TOM). The import of proteins into mitochondria is often mediated by heat shock proteins (e.g., HSP60 and HSP70), which specifically interact with a complex mitochondrial protein import apparatus, which includes matrix proteases. Physiological stimuli and stresses, including temperature changes and hormone treatment (e.g., thyroid hormone [TH]), may modulate the cardiac mitochondrial import apparatus pathways [18–20].
The number of proteins translocated from cytosol to the mitochondria, which include several of the proapoptotic proteins (e.g., Bax, Bid) and some protein kinases, seems to be increasing.
Mitochondrial Retrograde Signaling
Mitochondrial retrograde signaling is a pathway of communication from mitochondria to the nucleus that impacts many cellular activities under both normal and pathophysiological conditions. In both yeast and animal cells, retrograde signaling is linked to the mammalian (or mechanistic) target of rapamycin (mTOR) signaling, but its precise connections in cardiomyocytes have not yet been established. It has been suggested that inhibition of mTOR by rapamycin suppresses myocardial hypertrophy although its mechanism has not been found [21]. Song et al. [22] have recently studied the effect of mTOR on pathological hypertrophy and whether cardiac mTOR regulates the inflammatory response. Transgenic mice with cardiac-specific overexpression of wild-type mTOR (mTOR-Tg) were used. These mice were protected against the cardiac dysfunction induced by left ventricular pressure overload—aortic constriction (AC)—and showed lower levels of interstitial fibrosis than littermate controls (WT) at 4-week post-AC. In contrast, AC caused cardiac dysfunction in the WT. At 1-week post-AC, the proinflammatory cytokines interleukin (IL)-1β and IL-6 were significantly increased in WT mice but not in mTOR-Tg mice. This study further characterized the effects of mTOR activation, by exposing HL-1 cardiomyocytes transfected with mTOR to lipopolysaccharide (LPS). mTOR overexpression suppressed LPS-induced secretion of IL-6, and mTOR inhibitors rapamycin and PP242 abolished this inhibitory effect. Furthermore, mTOR overexpression reduced NF-κB-regulated transcription in HL-1 cells. Taken together, these findings suggest that mTOR protects against pressure-overload changes and its cardioprotective effect appears to be mediated by regulation of the inflammatory reaction [22]. The mTOR plays a critical role in the regulation of cell growth and in the response to energy state changes. Zhang et al. [23] have shown that ablation of Mtor in the adult mouse myocardium results in fatal, dilated cardiomyopathy (DCM) that is characterized by apoptosis, autophagy, altered mitochondrial structure, and accumulation of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). 4E-BP1 is an mTOR-containing multiprotein complex-1 (mTORC1) substrate that inhibits translation initiation. Mtor-ablated mice subjected to pressure overload showed impaired hypertrophic response and accelerated progression of HF. When the gene encoding 4E-BP1 was ablated together with Mtor, marked improvement was observed in apoptosis, heart function, and survival. These data demonstrate a role for the mTORC1 signaling network in myocardial response to stress and in particular highlight the role of 4E-BP1 in the regulation of cardiomyocyte viability and HF. Because the effects of reduced mTOR activity were mediated through increased 4E-BP1 inhibitory activity, blunting this mechanism may represent a novel therapeutic strategy to improve cardiac function in HF.
Role of Calcium in Nuclear-Mitochondrial Cross Talk
In mammalian cells, mitochondrial dysfunction sets off signaling cascades through altered Ca2+ dynamics including calcineurin activation, which activate several protein kinase pathways (e.g., PKC and MAPK) and transcription factors such as NF-κB, calcineurin-dependent NFAT, CREB, and ATF leading to stress protein expression (e.g., chaperone proteins) and activities [24]. This can result in both changes in cell morphology and phenotype including proliferative growth, apoptotic signaling, and glucose metabolism. Thus, calcium appears to be an important link between the relay of extracellular signals to the nucleus and the bidirectional communication between nucleus and mitochondria. Furthermore, this concept is strengthened by the observation that CREB is found in its transcriptionally active, phosphorylated state in cells exhibiting mitochondrial dysfunction resulting either from mtDNA loss or by pathological mutation in mtDNA [25], and this seems to happen simultaneously with activation of CaMKIV, leading to CREB phosphorylation in response to calcium. Nevertheless, whether calcium signaling through CREB, and the PGC-1 family coactivators, represents a physiologically significant pathway of retrograde regulation in vertebrate cells is not known [26].
Endoplasmic Reticulum
The endoplasmic reticulum (ER) is a multifunctional signaling organelle that contributes to the regulation of cellular processes such as the entry and release of Ca2+, sterol biosynthesis, apoptosis, and release of arachidonic acid [27]. One of its primary roles is to function as a source of the Ca2+ signals that are released through either IP3 or ryanodine receptors (RyRs), which are themselves Ca2+-sensitive. Another significant function of the cardiomyocyte ER is to regulate apoptosis by operating in tandem with mitochondria. In cells undergoing activation or excitation, calcium is released from the endoplasmic/sarcoplasmic reticulum to activate calcium-dependent kinases and phosphatases, thereby regulating numerous cellular processes, for example, apoptosis and autophagy [28]. Antiapoptotic regulators of apoptosis such as Bcl-2 may act by reducing the ebb and flow of Ca2+ through ER/mitochondrial cross talk [29]. Because both organelles are in close proximity, calcium is rapidly taken up by mitochondria via the calcium uniporter on the inner mitochondrial membrane [30–32]. Bcl-2 and Bcl-xL inhibit calcium redistribution to the mitochondria, thereby limiting calcium uptake [29, 33]. Also, it has been suggested that Bcl-2 inhibits calcium release through L-type channels, thereby preventing mitochondrial calcium uptake [34].
The capability of the ER in spreading signals throughout the cell, mediated by a process of Ca2+-induced Ca2+ release, is particularly important in the control of cardiomyocyte function. The role of ER as an internal reservoir of Ca2+ is coordinated with its role in protein synthesis since a constant luminal level of Ca2+ is essential for protein folding. In order to achieve this regulation, the ER also contains several stress signaling pathways that can activate transcriptional cascades to regulate the luminal content of the Ca2+-dependent chaperones responsible for the folding and packaging of secretory proteins.
Key Players in Mitochondrial Signaling
Nuclear Gene Activation
Nuclear-mitochondrial interactions depend on the interplay between numerous transcription factors (NRF-1, NRF-2, PPARα, ERRα, Sp1, and others) and members of the PGC-1 family of regulated coactivators (PGC-1α, PGC-1β, and PRC) [26]. These nuclear transcription modulators govern the expression of a wide array of mitochondrial proteins in response to diverse cellular stimuli and signals. For instance, nuclear transcription factors such as nuclear respiratory factors (NRFs) 1 and 2 are implicated in the activation of mitochondrial biogenesis [35, 36, 38]. These factors exert a direct effect on the synthesis of specific nuclear-encoded subunits of the mitochondrial respiratory enzymes as well as indirectly by upregulating levels of TFAM, involved in both mtDNA replication and transcription (see Fig. 8.1). In addition, a master transcription coactivator (PGC-1) activates directly or indirectly the expression of transcription factors NRF1 and NRF2 [37]. These transcriptional models serve as a framework to understand the integration of cell-signaling events with mitochondrial biogenesis and function [26].
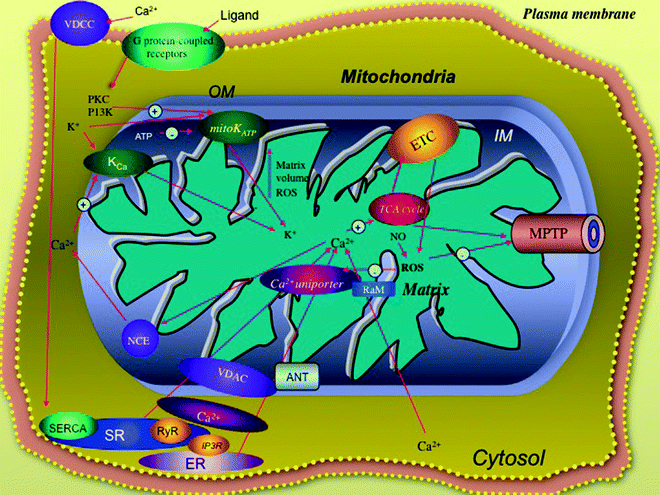
Fig. 8.2
Myocardial mitochondria ion channels are critical signal transducers. The cardioprotection pathway involving the mitochondrial K+ channels (mitoKATP) is located in the inner membrane (IM) and is mediated by reactive oxygen species (ROS), ligands binding to G protein-coupled membrane receptors, and protein kinases (PKC/PI3K). Calcium enters the mitochondria in response to stimuli via a complex of membrane proteins, including the outer membrane (OM) voltage-dependent anion carrier (VDAC), the IM proteins, the rapid mode of Ca2+ uptake channel (RaM), and the Ca2+ uniporter; high Ca2+ levels can be supplied from ryanodine receptor (RyR) and inositol trisphosphate receptors (IP3R) located in the sarcoplasmic (SR) and endoplasmic (ER) reticulum, respectively, and are coupled to the entry of calcium into the cardiomyocyte through the voltage-dependent Ca2+ channel (VDCC). Entry of high calcium levels into the mitochondria can increase activities of several enzymes of tricarboxylic acid (TCA) cycle, electron transport chain (ETC), and complex V. Calcium also modulates the opening of the mitochondrial permeability transition pore (MPTP). Ca2+ efflux is primarily managed by the Na+/Ca2+ exchanger (NCE). Other abbreviations: ANT adenine nucleotide translocator K Ca mitochondrial Ca2+-regulated potassium channel, PKC protein kinase C, PI3K phosphatidylinositol 3-kinase
The nucleus also contains global regulatory transcription factors such as PPARs and their transcriptional coactivators (i.e., PPAR, RXR) which also play a pivotal regulatory role in the expression of mitochondrial protein components of FAO pathway, integral to bioenergetic metabolism [38–40]. Transcriptional regulation by these activators is affected by hypoxia, ischemia, and HF [41, 42]. Expression of PGC-1 and mitochondrial biogenesis are modulated by activation of a calcium-/calmodulin-dependent protein kinase, indicating that the calcium-regulated signaling pathway plays a critical role in transcriptional activation of genes responsible for mitochondrial biogenesis [43]. Furthermore, these nuclear activators are modulated during cardiac development [44, 45]; activation of the nuclear-factor-activated T cell gene has been shown to be crucial in early cardiac development and is required to maintain myocardial mitochondrial oxidative function. Targeted cardiac gene disruption of nuclear-factor-activated T cell genes results in cardiomyocyte mitochondria ETC dysfunction, reduced ventricular size, and aberrant cardiomyocyte structure in mice embryo [45]. These nuclear transcriptional factors modulate the cardiac phenotype in transgenic animals exhibiting either mutations in specific transcriptional factors or overexpressed genes [37–45]. Also, it has been shown that specific nuclear transcription factors are essential for normal cardiac phenotype and mitochondrial function. For instance, cardiac-specific PGC-1 overexpression in transgenic mice results in uncontrolled mitochondrial proliferation and extensive loss of sarcomeric structure leading to dilated cardiomyopathy (DCM) [37]. Myocardial overexpression of PPAR may lead to severe cardiomyopathy with both increased myocardial fatty acid uptake and mitochondrial FAO [46]. In a similar manner, mutations targeting TFAM produce inactivation of myocardial mitochondrial gene expression and ETC dysfunction resulting in DCM and atrioventricular conduction defects [47].
Protein Kinases
Indications that mitochondria contain multiple phosphoprotein substrates for protein kinases and that a number of protein kinases are translocated into heart mitochondria strongly suggest that protein phosphorylation within the mitochondria is a critical element of mitochondrial signaling pathways [48]. However, it is important to note that detection of a protein in a phosphorylated state does not mean that such phosphorylation plays a regulatory role. Many proteins can be phosphorylated in vitro by protein kinases yet show no changes in activity. Up to this point, protein kinases identified in heart mitochondria include PDH kinase, branched-chain keto acid dehydrogenase kinase, PKA, PKC isoforms, and c-Jun N-terminal kinase [49–54], and their characterization has provided new insights into the fundamental mechanisms regulating mitochondrial responses to diverse physiological stimuli and stresses.
In cardiomyocytes, PKC after translocation to the mitochondria forms a signaling module by complexing with specific mitogen-activated protein kinases (e.g., extracellular signal-regulated kinase, p38, and c-Jun N-terminal kinase), resulting in phosphorylation of the proapoptotic protein Bad [53]. Also, PKC forms physical interaction with components of the cardiac mitochondrial permeability transition pore (MPTP) (in particular, VDAC and ANT) [55]. This interaction may inhibit the pathological opening of the pore (including Ca2+-induced opening and subsequent mitochondrial swelling) contributing to PKC-induced cardioprotection (CP). While PKC activation in CP likely precedes mitoKATP channel opening, its direct interaction with the mitoKATP channels has not been demonstrated. Following treatment with diazoxide, PKC is also translocated to cardiac mitochondria [52]; however, several observations have shown that PKC does not play a contributory role in the CP provided by ischemic preconditioning [56, 57].
A mitochondrial PKA (mtPKA) and its protein substrates have been localized to the matrix side of the inner mitochondrial membrane [51]. In cardiomyocytes the NDUFS4 is phosphorylated by mtPKA, and increased levels of cAMP promote NDUFS4 phosphorylation, enhancing both complex I activity and NAD-linked mitochondrial respiration [51, 58, 59]. These posttranslational changes can be reversed by dephosphorylation via a mitochondria-localized phosphatase. Phosphorylation of several subunits of COX, including COXI, III, and Vb, occurs at serine residues by mtPKA, modulates COX activity [60], and has been considered to be a critical element of respiratory control. This cAMP-dependent phosphorylation occurs with high ATP/ADP ratios and results in allosteric inhibition of COX activity; at the same time this regulatory control can result in reduced membrane potential and more efficient energy transduction in the resting state. On the other hand, increases in mitochondrial phosphatase (Ca2+-induced) reverse allosteric COX inhibition/respiratory control resulting in increased membrane potential and ROS formation; interestingly, various stress stimuli leading to increased Ca2+ flux result in increased potential and mitochondrial ROS formation. Newly developed kinase inhibitor assays and proteomic analysis have allowed identification of a variety of mitochondrial phosphoprotein targets for these kinases. A set of proteins has been found in the mitochondrial phosphoprotein proteome of bovine heart as protein targets of kinase-mediated phosphorylation [61]. The majority of these phosphoproteins were involved in mitochondrial bioenergetic function either in the TCA cycle (e.g., aconitase, isocitrate, and pyruvate dehydrogenase) as respiratory complex subunits (e.g., NDUFA 10 of complex I, succinate dehydrogenase flavoprotein subunit of complex II, core I and core III subunits of complex III, and subunits of complex V) or as essential players in the homeostasis of mitochondrial bioenergetics (e.g., creatine kinase, ANT). Interestingly, phosphorylation of the elongation factor Tu, a key regulatory protein of the cardiac mitochondrial protein translation apparatus, is modulated during myocardial ischemia [62].
Calcium Signaling
The import of Ca2+ from cytosol into cardiac mitochondria is an important regulatory event in cell signaling. The organelle couples the cellular metabolic state with Ca2+ transport processes not only modulating their own intraorganelle Ca2+ but also influencing the entire cellular network of cellular Ca2+ signaling, including the ER, the plasma membrane, and the nucleus [63]. These interactions are critical in Ca2+ homeostasis and signaling in the cardiomyocyte and in the numerous cellular functions regulated by the cation [64, 65]. Bidirectional signaling mediated by Ca2+ provides a framework by which mitochondrial biogenesis, structure, and metabolic activity will dictate a number of adaptive mechanisms during cell proliferation and cellular stress. PGC-1α selectively reduced mitochondrial Ca2+ responses to cell stimulation by reducing the efficacy of mitochondrial Ca2+ uptake sites and increasing organelle volume. In turn, this affected ER Ca2+ release and cytosolic responses in HeLa cells. Above all, the modulation of mitochondrial Ca2+ uptake significantly reduced cellular sensitivity to the Ca2+-mediated proapoptotic effect of C2 ceramide [66, 67]. Mitochondrial calcium flux, particularly in cultured cardiomyocytes, has become detectable using advanced cell imaging techniques with fluorescent dyes, confocal microscopy, and recombinantly derived Ca2+-sensitive photoprobes [68, 69]. Mitochondrial calcium influx is primarily provided by a Ca2+ pump uniporter (see Fig. 8.2) located in the inner membrane and driven by the mitochondrial membrane potential as well as by low matrix Ca2+ levels and can be blocked by ruthenium red [70].
Mitochondrial Ca2+ uptake is significantly and rapidly elevated in cardiomyocytes during physiological Ca2+ signaling and is often accompanied by a highly localized transient mitochondrial depolarization [69]. Efflux of Ca2+ from cardiomyocyte mitochondria is mediated by a Na+/Ca2+ exchanger (NCE) linked to ETC proton pumping, although calcium efflux also occurs with MPTP opening. Activation of the MPTP and mitochondrial Ca2+ flux also occur in early myocardial apoptosis and IRI and are involved in the generation of a calcium wave delivering system between adjacent mitochondria [71]. A major consequence of increased mitochondrial Ca2+ uptake is the upregulation of energy metabolism and stimulation of mitochondrial OXPHOS. Elevated mitochondrial Ca2+ levels allosterically stimulate the activity of three TCA cycle enzymes including pyruvate, isocitrate, and 2-oxoglutarate dehydrogenases [72, 73]. Activation of these enzymes by Ca2+ results in increased NADH/NAD+ ratios and ultimately leads to increased mitochondrial ATP synthesis. A thermokinetic model of cardiac bioenergetics showed calcium-dependent activation of the dehydrogenases as the rate-limiting determinant of respiratory flux regulating myocardial oxygen consumption, proton efflux, and NADH and ATP synthesis [74]. In cardiomyocytes mitochondrial ATP synthase activity can be directly modulated by increased mitochondrial Ca2+ levels [75, 76].
Many cellular processes require cooperation between the plasma membrane, the nucleus, and the subcellular vesicular/tubular networks such as the mitochondria and endoplasmic reticulum. Contacts between these cellular structures are crucial for the synthesis and intracellular transport of phospholipids as well as for intracellular Ca2+ homeostasis, controlling fundamental processes like motility and contraction, secretion, cell growth, proliferation, and apoptosis. Contacts between smooth subdomains of the endoplasmic reticulum and mitochondria have been shown to be required also for maintaining mitochondrial structure [63]. The small distance between the organelle membranes as measured by electron microscopy allows contact formation by proteins present on their surfaces, permitting and regulating their interactions. Intracompartment Ca2+ signaling is recognized as a key mode of signal transduction and amplification in mitochondria [68, 69]. Using product of phosphatidylinositol 4,5-bisphosphate hydrolysis, such as inositol trisphosphate (IP3), as second messengers, a variety of cell-surface hormones and neurotransmitters signal the release of Ca2+ from endoplasmic reticulum (ER) and Golgi apparatus to the cytosol. The proximity of mitochondria to ER membranes appears to be a significant factor for ER Ca2+ release and mitochondrial Ca2+ uptake [77]. This dramatic increase in mitochondrial Ca2+ is rapidly mobilized from the ER-IP3 receptor channels (Fig. 8.2) when in close contact to mitochondria, albeit the precise molecular mechanism of this transfer has not been fully established. Similarly, the sarcoplasmic reticulum ryanodine receptors are also located near the cardiomyocyte mitochondria undergoing calcium release [78]. Proposed mechanisms for the rapid calcium mitochondrial import include the involvement of diffusible cytosolic factors that stimulate the Ca2+ uniporter, activation of rapid mode of uptake (RaM), and enhanced uptake by mitochondrial analogues of ryanodine receptors residing in the inner membrane [79–82]. A voltage-dependent anion carrier has also been identified as a component in Ca2+ transport from ER through the outer mitochondrial membrane [83].
Mitochondrial KATP Channel
Besides their sarcolemmal location, KATP channels present in the inner membrane of mitochondria (mitoKATP) Fig. 8.2. These cardioprotective KATP channels consist of pore-forming (Kir6.1 and/or Kir6.2) and sulphonylurea-binding modulatory subunits [sulfonylurea receptor (SUR) 1, 2A, or 2B]. While KATP channels consisting of Kir6.2/SUR2A subunits present in heart muscle and Kir6.1/SUR2B subunit-containing KATP channels present in smooth muscle, the SUR1-containing channels have been found to participate in pancreatic insulin release. However, the SUR1 subunits have also been found recently in mouse cardiac tissue and show protection from myocardial IRI in SUR1-null mice.
The molecular identification and functional characterization of a mitochondrial sulfonylurea receptor 2 splice variant generated by intraexonic have been reported by Ye et al. [84] They found that isolation of transcripts encoding the 55-kDa forms showed a nonconventional intraexonic splicing (IES) event, which occurred in the SUR2 mRNA to generate such variants. Further investigation suggested these modified channels formed by IES variants generated a distinct ATP-sensitive current. This same group of investigators also reported that SUR2 knockout (SUR2KO) male mice are protected against IRI and that a 55-kDa intraexonic splice variant of SUR2A (SUR2A-55), unique to mitochondrial membrane fractions, forms functional KATP channels and is more abundant in mitochondria of SUR2KO mice than in wild type. Interestingly, overexpression of SUR2A-55 seems to reduce infarct size and maintains cardiac function with improved maintenance of the mitochondria membrane potential [85].
ATP-sensitive potassium channels of the inner mitochondrial membrane (mitoKATP) are blocked by ATP and have been implicated as potential mediators of cardioprotective mechanisms such as IPC [86]. This cardioprotective effect is partially mediated by attenuating Ca2+ overloading in the mitochondrial matrix and by increased ROS generation during preconditioning, further leading to protein kinase activation and decreased ROS levels generated during reperfusion [87]. The mitoKATP channel is also regulated by a variety of ligands (e.g., adenosine, opioids, bradykinin, acetylcholine), which bind sarcolemmal G protein-coupled receptors, with subsequent activation of calcium flux, tyrosine protein kinases, and phosphatidylinositol 3-kinase (PI3K)/Akt (protein kinase B) pathway [88, 89]. Marked changes in mitochondrial matrix volume associated with mitoKATP channel opening may play a contributory role in the CP process [89], although this has been questioned [90]. MitoKATP channel opening is specifically activated by drugs such as diazoxide and nicorandil and can also inhibit H2O2-induced apoptotic progression in cardiomyocytes, suggesting that mitoKATP channels may also play a significant role in mediating OS signals in the mitochondrial apoptotic pathway [91, 92]. Another ion channel (i.e., the calcium-activated K+ channel) has been identified on the mitochondrial inner membrane and has been shown to have a cardioprotective effect [93]. Nevertheless, the precise temporal order of events in the mitochondrial CP cascade and the exact molecular nature of the mitoKATP channel remain to be defined [94].
Mitochondrial Permeability Transition Pore
The MPTP is a protein pore formed in the mitochondria membranes under certain pathological conditions. The opening of this megachannel, located at contact sites between the inner and outer membranes, has been suggested to cause a number of important changes in mitochondrial structure and metabolism, including increased mitochondrial matrix volume (leading to mitochondrial swelling); release of matrix Ca2+; altered cristae; cessation of ATP production, primarily due to uncoupling of the ETC; and dissipation of the mitochondrial membrane potential [95]. The MPTP may be a site where the organelle integrates multiple cell-signaling stimuli as well as metabolic responses, and the induction of the MPTP can lead to cell death and play a critical role in apoptosis. Interestingly, MPTP opening during apoptosis is transient, allowing mitochondria to maintain the ATP levels necessary for fueling the downstream apoptotic responses. It should not be surprising that early apoptotic events involve the modulation of ATP levels, given the close proximity of the MPTP to the respiratory complexes in the mitochondrial inner membrane as well as its involvement in the mitochondrial loss of cytochrome c, a critical molecule in ETC function. Present at the MPTP site are a number of energy-associated mitochondrial molecules including the ANT, the glycolytic enzyme hexokinase, the outer membrane voltage-dependent anion channel protein (VDAC or porin), the mitochondrial creatine kinase, and the phospholipid cardiolipin [96]. While ample data exist on MPTP subunit composition, genetic knockout experiments suggest that the identity of the core factors of the MPTP is still unresolved, and gathered evidence suggests a much more complex composition of this protein complex than anticipated [97].
Following an episode of myocardial ischemia, at the onset of reperfusion, opening of this nonspecific pore is a critical determinant of myocyte death. Besides its role in the mitochondrial pathway of apoptosis, the opening of the MPTP, if unrestrained, leads to the loss of ionic homeostasis and ultimately to necrotic cell death [98]. Fatty acids, high matrix Ca2+ levels, pro-oxidants, metabolic uncouplers, NO, and excessive mitochondrial ROS production (primarily from respiratory complex I and III) promote the opening of the MPTP.
Moreover, the MPTP may be also an important target of CP. It has been shown that suppressing MPTP opening at the onset of reoxygenation can protect human myocardium against lethal hypoxia-reoxygenation injury [99] and that the inhibition of MPTP opening can be mediated either directly by cyclosporin A (CsA) and sanglifehrin A (SfA) or indirectly by decreasing calcium loading and ROS levels (Fig. 8.3).
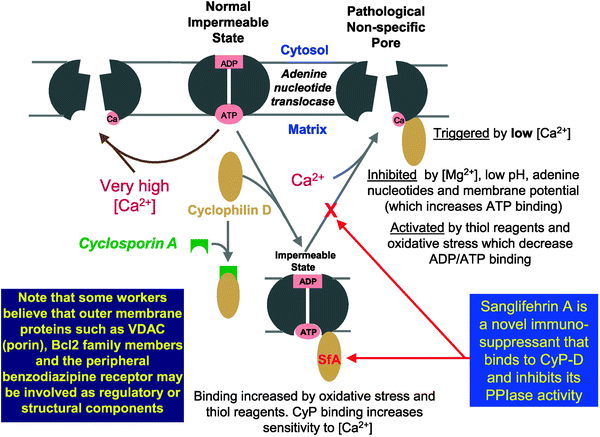
Fig. 8.3
The proposed molecular mechanism of the mitochondrial permeability transition pore (from Halestrap et al. [97] with permission from Elsevier)
Recently, Penna et al. [100] reported that very low concentrations (pmol/L) of platelet-activating factor (PAF) infused before ischemia induce CP similar to those afforded by ischemic preconditioning (IP) and that endogenous PAF production participates in the induction of IP itself. This IP-like action exerted by low concentrations of PAF is due to the activation/phosphorylation of kinases included in the reperfusion injury salvage kinase (RISK) pathway, such as protein kinase C, Akt/PKB, and nitric oxide synthase. Together with the activation of mitochondrial KATP channels, these events may allow prevention of MPTP opening at reperfusion. In addition, the nitric oxide-dependent S-nitrosylation of L-type Ca2+ channels induced by PAF reduces intracellular Ca2+ overload.
Survival and Stress Signals Impact Heart Mitochondria
The list of extracellular influences and intracellularly generated signals which impact the mitochondrial organelle is growing, as reflected in Table 8.1. In addition to hormonal and cytokine stimuli (e.g., TH, TNF-α, interleukins), there are also pro/antiapoptotic modulators, nutrients, serum, growth and mitotic factors, as well as stress and metabolic stimuli which we describe in more detail in this section.
Table 8.1
Stimuli signaling myocardial mitochondrial function
Stimulus | Signaling pathway | Cardiac myocyte phenotype | Mitochondrial effect | Reference |
|---|---|---|---|---|
IL-1b | NO production | Cardiac dysfunction | Decreased respiration | [101] |
TNF-α | Ceramide pathway | Cell death | Reduced activity levels of PDH, complex I and II | [102] |
Heat stress | Increased levels of HSP 32,60,72 | Improved cardiac function after IRI | Increased complex I–V activities | [103] |
Low glucose | Myocardial apoptosis | Cell death | Cyt c release | [104] |
Low serum | Myocardial apoptosis | Cell death | Cyt c release | [104] |
Palmitate | Myocardial apoptosis; ceramide increase | Cell death | Reduced complex III and membrane potential; increased cyt c release, UCP and swollen mitochondria | [105] |
Ceramide | Ceramide pathway | Cell death | Decreased complex III activity | [106] |
Electrical stimulation | NRF-1 activation | Hypertrophy | Mitochondrial proliferation | [91] |
Nitric oxide | Peroxynitrite formation | Myocardial O2 uptake decline; increased H2O2 | Complex I and IV decrease; increased cyt c release | |
Thyroid hormone (T3/T4) | Receptor-mediated nuclear and mtDNA gene activation | Hypertrophy | Mitochondrial proliferation; increased UCP and uncoupled OXPHOS |
Survival Signals
The PI3K/Akt pathway promotes cell survival primarily by intervening in the mitochondrial apoptosis cascade at events before cytochrome c release and caspase activation occur (Fig. 8.4). Akt activation inhibits changes in the inner mitochondrial membrane potential that occur in apoptosis (suppressing apoptotic progression and cytochrome c release induced by several proapoptotic proteins). While Akt also contributes to the phosphorylation and inactivation of the proapoptogenic protein Bad, it remains unclear whether Bad phosphorylation is the mechanism by which Akt ensures cell survival and mitochondrial integrity since other mitochondrial targets of Akt remain to be identified [111]. Interestingly, microarray analysis has demonstrated that once unnoticed Akt regulation of genes could modulate the influence of Akt on cardiomyocyte survival, metabolism, and growth [112].
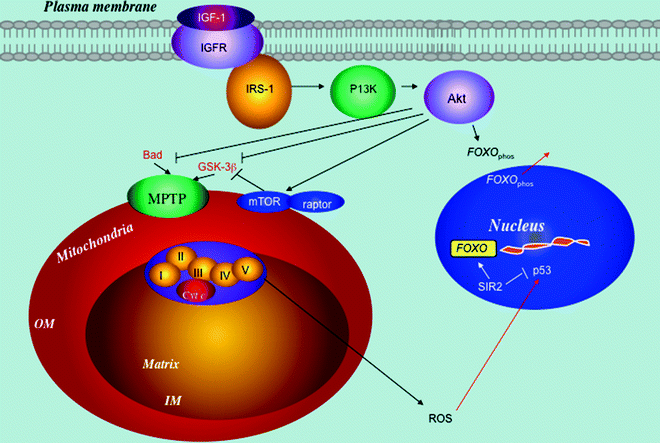
Fig. 8.4
The myocardial phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway has a mitochondrial component. Interacting signaling pathways, including insulin-like growth factor (IGF), mammalian target of rapamycin (mTOR), and sirtuin (SIR) 2, are shown. Downstream of the receptor, the signal is transmitted to kinases (first PI3K and later Akt). Activation of Akt results in the negative regulation of proapoptotic protein (e.g., Bad), apoptotic suppression, and upregulation of glucose uptake. It also attenuates glycogen synthase kinase-3β (GSK-3β) activity-reducing mitochondrial permeability transition pore (MPTP). Furthermore, Akt activates mTOR signaling, part of the mitochondrial retrograde pathway. Akt phosphorylates forkhead box protein O (FOXO), inactivating it and increasing its translocation from the nucleus to the cytosol. On the other hand, the SIR2 activates FOXO transcriptional activity by reversing its acetylation. Similarly, SIR2 inactivates p53 by deacetylation and attenuates its apoptotic program. Other abbreviations: Cyt c cytochrome c, IGFR insulin-like growth factor 1 receptor, IM mitochondrial inner membrane, IRS insulin receptor substrate, OM mitochondrial outer membrane, raptor regulatory-associated protein of mTOR, ROS reactive oxygen species
Deprivation of nutrients (e.g., amino acids), glucose, and growth factor, which can lead to cardiomyocyte apoptosis [104], has been found to signal via the mitochondria-associated mTOR protein (Fig. 8.4) [113]. Moreover, both the Akt pathway and the downstream mTOR protein impact cardiomyocyte survival and cell size largely through increased cytoplasmic protein synthesis by mediating activation of translational initiation factors and ribosomal proteins. In addition, serotonin binding to the serotonin 2B receptor protects cardiomyocytes against serum deprivation-induced apoptosis via the PI3K pathway impacting ANT and Bax expression. Transgenic mice harboring serotonin 2B receptor null mutations manifest pronounced myocardial mitochondrial defects in addition to altered mitochondrial ETC activities (complex II and IV), ANT-1, and Bax expression [114].
Finally, Akt signaling also provides CP against ischemic injury in response to diverse treatments including cardiotrophin-1, acetylcholine, adenosine, and bradykinin-mediated preconditioning [115–117], although the precise target of Akt action in mitochondria-based cardioprotection remains undetermined since Akt does not associate directly with mitoKATP channels.
Stress Signals
Stresses in cardiac hypertrophy (e.g., mechanical) and ischemia/hypoxia (e.g., oxidative) elicit a variety of adaptive responses at the tissue, cellular, and molecular levels. A model showing the cardiac physiological response to hypoxia reveals that mitochondria function as O2 sensors both by increasing ROS generation during hypoxia and via their abundant heme-containing proteins (e.g., COX), which reversibly bind oxygen [118]. Oxidant signals such as ROS act as second messengers initiating signaling cascades and are prominent features in both adaptive responses to hypoxia and mechanically stressed heart. Downregulation of COX activity contributes to the increased ROS generation and signaling observed in cardiomyocytes during hypoxia [119]. Also, hypoxia stimulates NO synthesis in cardiomyocytes [120], and NO downregulates COX activity with subsequent mitochondrial H2O2 production. This event has been proposed to provide a mitochondria-generated signal for further regulating redox-sensitive signaling pathways, including apoptosis, and can proceed even in the absence of marked changes in ATP levels [108]. Interestingly, nitric oxide synthase has been identified in heart mitochondria although its role in regulating OXPHOS is not clear [121, 122]. Mitochondrial ROS has also been shown to activate p38 kinase in hypoxic cardiomyocytes [123]. Longer-term responses to hypoxia have been shown to involve increased gene expression of hypoxia-induced factors and the activation of transcription factors such as NF-κB which has also been implicated in the complex regulation of cardiac hypertrophy and inflammatory cytokines (TNF-α, interleukin-1). While increased ROS has been shown to be an important element in NF-κB gene activation, there is recent evidence showing that cardiomyocyte hypoxia-induced factor gene activation can also occur in the absence of ROS [124].
Metabolic Signals and UCPs
When cells need thermal energy, they employ special protein(s) that do not allow the proton gradient potential to be used by the ATP-making machinery; instead it is transformed into heat. These proteins are known as uncoupling proteins (UCPs) since they uncouple the ATP-making process from the proton pump (Fig. 8.5). Also, shown in Table 8.2 are UCPs hitherto known.
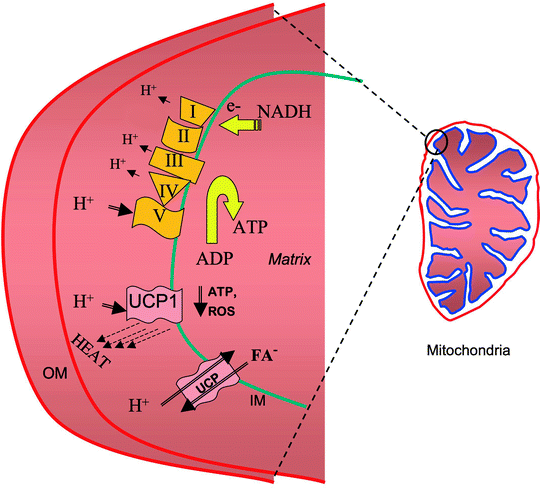
Fig. 8.5
Schematic representation of UCP’s uncoupling mechanisms. Uncoupling proteins (UCPs) uncouple the ATP-making process from the proton pump. Abbreviations: FA fatty acid, IM mitochondrial inner membrane, OM mitochondrial outer membrane, ROS reactive oxygen species
Table 8.2
Uncoupling proteins homologues
Protein | Presence and abundance |
|---|---|
UCP1 | Brown adipose tissue |
UCP2 | Macrophages, pancreatic β-cells, skeletal muscle |
UCP3 | Heart, skeletal muscle |
UCP4 | Nervous system |
UCP5 (BMCP1) | Brain |
KMCP1 | Kidney |
Mitochondrial UCPs are members of a family of mitochondrial anion carrier proteins (MACP). They are nuclear-encoded transmembrane transporter proteins located in the mitochondrial inner membrane [125]. UCP1, mainly expressed in brown adipose tissue (BAT), was the first to be discovered and is responsible for thermogenesis in animals. UCP2 is originally thought to play a role in nonshivering thermogenesis, obesity, and diabetes; its main function appears to be in the control of mitochondria-derived ROS. Another uncoupling protein homologue, the UCP3, is mainly expressed in skeletal muscle and BAT, and its gene is transcribed from tissue-specific promoters in humans but not in rodents. All the members of this protein family possess a common feature of shunting protons across the mitochondrial inner membrane and reduce ATP synthesis; however, this common mechanism of action is used to carry out different functions by the different UCPs. These proteins underscore the new roles played by mitochondria, besides their function in ATP production, apoptosis, OS damage, diverse cardiac pathologies, and aging. For example, UCPs are involved or affected in IRI and HF as shown in Fig. 8.6.
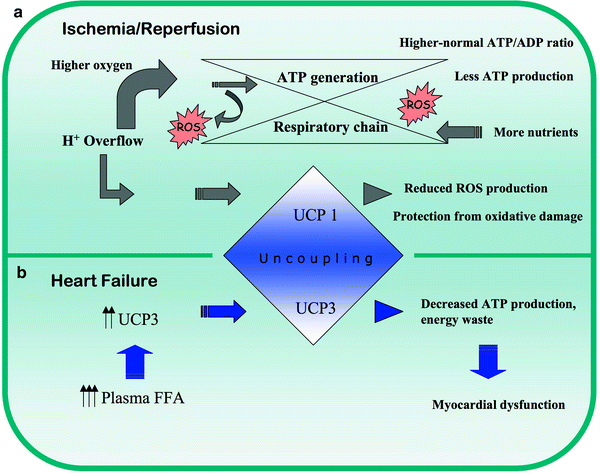
Fig. 8.6
Response of uncoupling proteins in (a) ischemia/reperfusion and (b) heart failure. FFA free fatty acids, ROS reactive oxygen species, UCP uncoupling protein 1/2/3
New discoveries are advancing our understanding of UCP’s roles in cardiovascular physiology. These mitochondrial inner membrane-localized carrier proteins function to dissipate the proton gradient across the membrane. Expression of uncoupling proteins is upregulated transcriptionally with either palmitate or TH treatment [126–128]. Interestingly, cardiac expression of one of the uncoupling protein genes (UCP3) has also been reported to be PPAR dependent [128]. In addition, increased expression of uncoupling proteins in cardiac muscle results in increased uncoupling of OXPHOS from respiration, decreased myocardial efficiency, and mitochondrial membrane potential [127]. Potential biochemical and physiological processes where UCPs are involved or affected are presented in Fig. 8.7. Upregulation of UCP 2 and 3 mRNA expressions in human skeletal muscle mitochondria by TH occurs without coordinated induction of respiratory chain genes. Using a whole animal/whole organ-heart model, Barbe et al. [129] administered TH to Wistar rats for 7 days. Within 24 h after the last dose, heart mitochondria were isolated, and UCP levels were determined. UCP2 and UCP3 increased by about 40%, and mitochondrial uncoupling, as measured by oligomycin-insensitive respiration rate, increased twofold in the presence of palmitate. In the isolated working heart, the presence of palmitate significantly reduced cardiac output and efficiency by about 36% in the TH-treated rats [127]. Thus, increased UCPs in hyperthyroid rats are associated with increased uncoupling and decreased myocardial efficiency in the presence of palmitate.
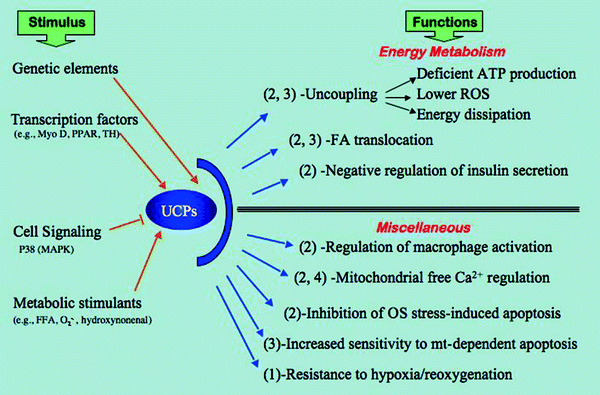
Fig. 8.7
Flow chart showing an overview of uncoupling protein functions. The chart enlists the effectors as “stimulus” for uncoupling protein (UCP) activity and the resulting activities as “function” of UCPs. Number in parenthesis indicates the specific UCP 1, 2, 3, or 4 related to that activity. Abbreviations: FA fatty acid, FFA free FA, MAPK mitogen-activated protein kinase, Myo D myogenic differentiation antigen, Mt mitochondria, O 2•− superoxide anion, OS oxidative stress, P38 38-kDa MAPK, PPAR peroxisome proliferator-activated receptor, ROS reactive oxygen species, TH thyroid hormone
In muscle cells UCP3 increases FAO and decreases ROS [130]. In L6 muscle cells, when compared to the effect of an uncoupler agent like dinitrophenol, UCP3 seems to preferentially promote FAO utilization rather than glucose. Also, UCP3 reduces ROS production without significant increase in oxygen consumption. This function of UCP3 may be particularly useful in managing type 2 diabetes where impaired fatty acid metabolism and ROS handling set the stage for muscular insulin resistance.
Interestingly, UCP1 confers resistance against hypoxia/reoxygenation in a specific heart cell line [131]. H9c2 cell line transfected with UCP1 showed that overexpression of UCP1 was not compromising cell viability. In hypoxia/reoxygenation experiments, compared to control cells, the UCP1-expressing cells show a moderate decrease in their OXPHOS capacity but significantly higher survival with largely preserved subcellular ultrastructure. In these cells the surge in ROS production was significantly reduced.
UCP2 from the mitochondrial matrix side is activated by superoxide. Also, reactive aldehydes, such as 4-hydroxy-2-noneal produced from peroxidation of membrane phospholipids, as a result of oxidative damage, induce mitochondrial uncoupling through UCPs [132–134].
In cardiomyocytes under OS induced by 100 microM/L H2O2, UCP2 overexpression inhibits the mitochondrial death pathway. Furthermore, early apoptotic events (i.e., decrease in membrane potential), increased ROS generation, Ca2+ overload, and late-phase apoptotic events as detected by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) and caspase-3 activity have been assessed in cells transfected with an adenoviral vector containing human UCP2. UCP2 overexpression significantly suppressed the apoptotic events caused by OS secondary to H2O2 exposure. These data suggest that UCP2 may mitigate cell death occurring in ischemia/reperfusion injury by preventing apoptotic events, through preserving membrane potential and lowering ROS production [135].
Hypoxia decreased UCP3 expression in rat cardiomyocytes without changing mitochondrial respiratory coupling, while UCP2 expression was unchanged. A role for UCP3 in the regulation of heart FAO has been proposed instead of a role in uncoupling mitochondria. Hypoxia-induced regulation indicates a distinct mitochondrial regulatory function in the heart in response to metabolic stress [136]. Hypoxia as well as exercise increases rat skeletal muscle UCP3 levels by four- to sixfold compared to controls. Furthermore, AMPK, known to be stimulated during exercise and hypoxia, induces UCP3 expression [137]. These observations present further evidence supporting UCP tissue-specific behavior. Finally, the role that UCP2 and UCP3 play in cardiac pathophysiology appears to be dependent on the surrounding conditions. For example, in ischemia preconditioning UCPs support the survival of heart tissue by playing the crucial role of preventing oxidative damage. On the other hand, in the presence of elevated levels of circulating fatty acid, disruption in energy metabolism is actually worsened by uncoupling with depletion of ATP, as it may occur in the diabetic heart. In these circumstances, the question of “is UCP a friend or a foe?” seems appropriate; however, to answer this question, further research is warranted.
Cardiomyopathy and Mitochondrial Signaling Defects
Specific mtDNA mutations with associated mitochondrial respiratory dysfunction have been reported in cardiomyopathies as well as in systemic encephalomyopathies with cardiac involvement. The latter includes Leigh disease, MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes), and MERRF (myoclonic epilepsy and ragged-red fibers) [138]. In addition, depletion of cardiac mtDNA levels concomitant with myocardial mitochondrial respiratory dysfunction has been found in patients with both dilated and hypertrophic cardiomyopathy and in patients and animal models treated with zidovudine [139, 140]. While the dependence of cardiac homeostasis on functional mitochondria is primarily attributed to needed ATP derived from OXPHOS for maintaining myocardial contractility, the role of cardiac mitochondria in responding to a variety of intracellular and extracellular signals, metabolic substrates, and physiological stresses has aroused increasing interest. Analysis of cardiomyocytes containing specific pathogenic mtDNA point mutations and deletions or depleted mtDNA levels, for their effect(s) on mitochondrial signaling, may provide further information on the role of mitochondrial cytopathy in cardiac disease pathogenesis.
Mutations at several different nuclear gene loci have been reported together with OXPHOS deficiencies in cardiomyopathies associated with Leigh syndrome, cytochrome c oxidase deficiency, and Friedreich ataxia [141–143]. Mutations in nuclear genes involved in mitochondrial biogenesis which contribute to cardiac disease-associated mitochondrial enzyme and mtDNA defects (including large-scale mtDNA deletions and mtDNA depletion) have been also reported [144]. As previously noted, involvement of both genomes in mitochondrial biogenesis and mitochondria-based pathogenesis is an important rationale to study the cross talk and regulatory signaling between both genomes as well as to increase the search for mutations involved in mitochondrial biogenesis and in the regulation of cardiac mitochondrial function. Mutations and physiological insults targeting various mitochondrial pathways (other than mitochondrial OXPHOS function) are contributory to cardiac disease. Defects in mitochondrial carnitine, fatty acid transport, and FAO have a crucial role in cardiac sudden death, bioenergetic dysfunction, cardiac dysrhythmias, and cardiomyopathy [138]. Transgenic mice with disruption of specific nuclear genes encoding mitochondrial proteins may lead to cardiomyopathy and HF. Ablation of genes (i.e., knockout mutations) encoding ANT, Mn-SOD, the frataxin gene associated with Friedreich ataxia, and the mitochondrial transcription factor A (TFAM) affect the cardiomyopathy phenotype and HF [145–147]. These findings emphasize the importance of mitochondria in the maintenance of a functional cardiac phenotype, mainly the mitochondrial role in cardiomyocyte apoptosis (i.e., ANT is an essential component of the MPTP, which mediates early apoptotic progression) and mitochondrial antioxidant response (e.g., Mn-SOD) to OS, a critical element of myocardial ischemia and mitochondrial cytoprotection.
Mitochondrial Signaling in Myocardial Ischemia and Cardioprotection
Since a comprehensive discussion on the mitochondria role in ischemia and cardioprotection will be presented in Chap. 16, here it will suffice to say that mitochondrial ETC flux and OXPHOS decline when the supply of oxygen is disrupted such as occurs in myocardial ischemia. The high-energy phosphates pool is rapidly depleted, pyruvate oxidation decreases, and ATP production is impaired. Hydrolysis of glycolytically derived ATP, and the resulting accumulation of lactate and pyruvate, leads to intracellular acidosis (which has a deleterious effect on cardiac function) and to the accumulation of myocardial sodium and calcium. Moreover, the energy deficit occurring as a result of ATP depletion is compounded by the deployment of ATP to reestablish a disturbed myocardial ionic balance, rather than to fuel contraction.
AMP and other intermediates accumulate while mitochondrial swelling and degeneration occur. In addition, the activity levels of respiratory complexes IV and V decrease during myocardial ischemia, leading to an increased level of mtDNA deletions [146, 147]. While sustained myocardial ischemia leads to ATP depletion and necrotic cell death, there is also evidence that both ischemia and hypoxia can activate cardiomyocyte mitochondrial death pathway with the opening of the MPTP together with mitochondrial membrane depolarization, eventual disruption of the mitochondrial membranes, and release of cytochrome c [148].
Mitochondrial matrix swelling can result in improved mitochondrial energy production with activated fatty acid oxidation, mitochondrial respiration, and ATP production [149]. A known effect of mitoKATP channel opening is the preservation of normal low outer membrane permeability to nucleotides and cytochrome c, beneficial effects abrogated by the mitoKATP channel inhibitor 5-HD [150]. During ischemia opening of the mitoKATP channels can also contribute to the tight structure of the intermembrane space, which is needed to preserve low outer membrane permeability to adenine nucleotides. Opening of the mitoKATP channel (Fig. 8.8) is considered both a signaling trigger and a mediator/effector of the cardioprotection (CP) pathway [150].
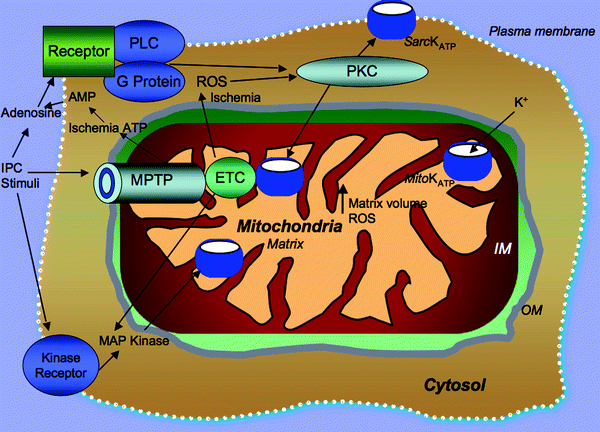
Fig. 8.8
Mitochondrial events occurring in early ischemic preconditioning. Adenosine, produced by the breakdown of adenine nucleotides during ischemia, leads to the activation of phospholipase C (PLC) which in turn activates and translocates protein kinase C (PKC) to mitochondrial membranes, where PKC modulates the opening of mitochondrial ATP-sensitive potassium channel (mitoKATP) and (to a lesser extent) sarcolemmal KATP channels. Other agonists of G protein-coupled receptors also stimulate parallel signaling pathways by stimulating either PKC-dependent or tyrosine kinase-dependent cascades which can confer cardioprotection (CP). Multiple mitochondrial events integral to CP include increased mitochondrial reactive oxygen species (ROS) generation, modulation of mitochondrial calcium intake, mitochondrial permeability transition pore (MPTP) opening, and mitochondrial electron transport chain (ETC) activity. The opening of the mitoKATP also provides positive feedback by affecting ROS, mitogen-activated protein kinase (MAP kinase), or PKC activation. Other abbreviations: IM mitochondrial inner membrane, IPC ischemic preconditioning, OM mitochondrial outer membrane, Sarc sarcolemma
Adenosine and its G protein-coupled receptors represent an important positive stimulus as well as a locus for feedback control of ischemic preconditioning (IPC) [151]. Adenosine is generated at high levels from ATP metabolism during myocardial ischemia, primarily in the mitochondrial organelle. IPC-induced CP has been found to be abrogated by treatment with adenosine receptor antagonists, suggesting that adenosine produced during IPC acts on cell-surface receptors [152]. Other ligands including opioids, bradykinin, acetylcholine, and endothelin, which bind specific G protein-coupled receptors, have been identified; they mediate both mitoKATP channel opening and elicit IPC-CP. These receptors (at least three surface receptors acting in parallel can trigger preconditioning) represent parallel and redundant pathways [153]. Also, recently Yang et al. [154] noted that during the trigger phase of IPC, adenosine, bradykinin, and opioid receptors are occupied, and although these three receptors trigger signaling through divergent pathways, the signaling converges on PKC. They proposed that at the end of the index ischemia the activated PKC sensitizes the low-affinity A(2b) adenosine receptor (A(2b)AR) through phosphorylation of either the receptor or its coupling proteins so that A(2b)AR can be activated by endogenous adenosine released by the previously ischemic cardiomyocytes. The sensitized A(2b)AR would then be responsible for activation of the survival kinases including PI3 kinase, Akt, and ERK which then act to inhibit lethal MPTP formation which normally uncouples mitochondria and destroys many myocytes in the first minutes of reperfusion. A marked reduction in infarct size in rats following administration of the selective A(2b)AR agonist BAY 60-6583 has been observed. While supportive of its concept as a cardioprotective receptor, recent observations indicated that the mechanism of the early phase of IPC is not dependent on signaling by the A(2b)AR [155]. A(2b)AR may contribute to the later stages of IPC dependent on the induction of stress-responsive genes.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


