Essentials of Diagnosis
- Dyspnea on exertion and at rest, orthopnea, paroxysmal nocturnal dyspnea, and fatigue.
- Elevated jugular venous pressure, basilar rales or coarse rales throughout both lung fields with wheezing, cardiomegaly, S3 gallop, hepatomegaly, and bilateral peripheral pitting edema.
- Dilated left ventricle with a reduced ejection fraction on transthoracic echocardiography.
- Elevated left ventricular filling pressures on cardiac catheterization.
General Considerations
Heart failure (HF) is a complex clinical syndrome resulting from any structural or functional myocardial dysfunction that leads to an impaired ability to circulate blood at a rate sufficient to maintain the metabolic needs of internal organs and peripheral tissues. The myocardial dysfunction is often the consequence of long-standing ischemia due to coronary artery disease or loss of myocardial mass due to prior infarction, long-standing myocardial stress due to suboptimally treated hypertension or valvular disease, or direct long-term toxin exposure (alcohol abuse, illicit substance use, or chemotherapeutic agents); rarely, fulminant infections (especially viral) can lead to autoimmune myocardial damage, and in some cases, there is no apparent cause (idiopathic cardiomyopathy, usually related to inherited or spontaneous gene mutations).
The cardinal manifestations of HF are dyspnea and fatigue (which may limit exercise tolerance) and fluid retention (which may lead to pulmonary/hepatic/splanchnic congestion and peripheral edema). Both abnormalities impair the functional capacity and quality of life of affected patients, but they may not necessarily dominate the clinical picture at the same time.
The prevalence of HF in the United States is around 5.8 million patients, of which approximately 45% have reduced ejection fraction/systolic dysfunction. There are over half a million cases of HF being newly diagnosed every year, and this incidence is expected to rise to over 700,000 new cases per year by 2040, mainly due to the aging of the population, as HF incidence doubles with each successive decade above age 45 for both men and women.
At age 40, the lifetime risk for HF is as high in women as in men, and is approximately one in five. Although over 75% of HF patients have antecedent hypertension or coronary artery disease, the population attributable risk (PAR) is different in men and women; the PAR for HF associated with coronary artery disease is 39% in men and 18% in women, whereas the PAR associated with hypertension is 39% in men and 59% in women, emphasizing the crucial need for population-based strategies to prevent the development of HF.
Multiethnic epidemiologic studies showed that the highest risk for developing HF is in African Americans (highest in young women and middle-aged men), followed by Hispanics/Latinos, Caucasians, and Chinese. This higher risk likely reflects differences in the prevalence of hypertension and diabetes mellitus and in socioeconomic status between different ethnicities.
Although the survival after HF diagnosis has improved, the 5-year mortality is still close to 50%, and over a quarter million deaths every year have antecedent HF. HF is one of the leading causes of hospitalization in the United States, with over one million hospitalizations a year (a figure that has not changed over the past decade). Indeed, 85% of patients diagnosed with HF will be hospitalized within 5 years of diagnosis, and over 40% will be hospitalized at least four times. HF accounts for over 12 million office visits and 6.5 million hospital days each year in the United States, with an estimated total cost of $39 billion, making it a major contributor to total health-care expenditure and the number one health expenditure for Medicare.
Pathophysiology
When an excessive workload is imposed on the heart by increased systolic blood pressure (pressure overload, such as in chronic hypertension or aortic stenosis), increased diastolic volume (volume overload, such as in progressive dilated idiopathic cardiomyopathy or chronic aortic or mitral regurgitation), or loss of myocardium (acutely in the setting of myocardial infarction, or chronically in the setting of flow-limiting coronary artery disease), normal myocardial cells hypertrophy in order to increase the contractile force of the unaffected areas. The biochemical, electrophysiologic, and contractile changes that ensue lead to alterations in the mechanical properties of the myocardium. Eventually, the compensatory force of the normal myocardial contraction decreases as cell loss and hypertrophy continue, leading to increased ventricular volume (“cardiac remodeling”) and significant geometric ventricular alterations (ellipsoid to spherical shape). After the initial compensatory phase, the increase in ventricular volume is associated with further reductions in the ejection fraction (progressive systolic dysfunction) and with abnormalities in the peripheral circulation from activation of various neurohormonal compensatory mechanisms.
In the United States, coronary artery disease is the underlying etiology for systolic HF in more than half of the cases and a major contributor in another 15–20%. Transient ischemic events may cause prolonged systolic dysfunction that persists even after the ischemic event has resolved (myocardial stunning). The sustained reduction in the coronary blood flow leads to tissue hypoperfusion that is sufficient to maintain viability, but insufficient to maintain a normal contractility (myocardial hibernation). However, hibernation cannot be maintained indefinitely, and myocardial necrosis will ensue if coronary blood flow is not restored. Ischemia and hibernation may lead to myocyte apoptosis, which may result in progression of left ventricular systolic dysfunction characterized by a reduction in cardiac output and/or an increase in wall stress.
These functional and structural myocardial changes result in compensatory activation of neurohormonal systems, such as the renin–angiotensin–aldosterone system (RAAS), the adrenergic system, and the hypothalamic–neurohypophyseal system. The mechanisms for RAAS activation in HF include renal hypoperfusion with decreased filtered sodium reaching the macula densa and decreased stretch of the juxtaglomerular apparatus leading to increased renin release. Renin cleaves four amino acids from circulating angiotensinogen to form the biologically inactive angiotensin I. Angiotensin-converting enzyme (ACE) cleaves two amino acids from angiotensin I to form the biologically active angiotensin II, which binds to vascular angiotensin receptors causing vasoconstriction of the efferent postglomerular arterioles (promoting reabsorption of sodium, urea, and water) and to cardiac angiotensin receptors causing myocyte hypertrophy, apoptosis, and interstitial fibrosis. Increased circulating levels of angiotensin II also promote the release of aldosterone from the adrenal zona glomerulosa, resulting in sodium reabsorption and potassium excretion in the distal nephron, along with myocardial fibroblast proliferation and collagen deposition. The abnormal hypertrophy and fibrosis increase the passive stiffness of the ventricles and arterial bed, interfering with ventricular filling and reducing arterial compliance and, along with myocyte slippage and interstitial growth, result in ventricular remodeling. Finally, angiotensin II may increase the release of arginine vasopressin via a mechanism that does not rely on changes in osmolality, leading to renal free water reabsorption and dilutional hyponatremia seen with severe HF.
In patients with HF, the inhibitory input from high- pressure carotid sinus and aortic arch baroreceptors and low-pressure cardiopulmonary mechanoreceptors decreases, while nonbaroreflex peripheral chemoreceptors and muscle metaboreceptors excitatory input increases, with a net increase in sympathetic nerve traffic and blunted parasympathetic nerve traffic, and a resultant loss of heart rate variability and increased peripheral vascular resistance. As a result of the increase in sympathetic tone, there is an increase in circulating levels of norepinephrine from a combination of increased release from adrenergic nerve endings and its consequent “spillover” into the plasma, with reduced uptake of norepinephrine by adrenergic nerve endings. In patients in early stages of HF, norepinephrine increases heart rate and contractility and may support cardiac function. However, in later stages of HF, cardiac response to norepinephrine is blunted, partly because of the decreased β-receptor density seen in patients with chronic severe HF, where a shift occurs from the normal density (β1/β2 ratio of 70–80%/20–30%) to a more even distribution of 60%/40%. In addition, α-adrenergic receptors increase in the failing heart so that the end result is a more balanced distribution of myocardial α, β1, and β2 receptors. Chronic adrenergic stimulation has been shown to induce expression of proinflammatory cytokines, such as tumor necrosis factor-α, interleukin-1, and interleukin-6. Increased cytokine levels may result in the skeletal muscle myopathy characteristic of HF and cause myocardial inflammation, cell proliferation, and apoptosis, thereby worsening the HF syndrome. Tumor necrosis factor-α also activates transcription factors and enzymes involved in signal transduction, and it induces a number of genes, including the fetal gene program. Peripheral arteriolar tone is also increased in patients with HF (via α-adrenergic receptor stimulation), producing an increase in blood pressure and ventricular afterload and vasoconstriction of the renal efferent arterioles that causes significant sodium and water retention and increases production of renin, angiotensin II, and aldosterone, emphasizing the tight interplay between the adrenergic system and the RAAS. Indeed, ACE inhibition in patients with HF has been found to reduce peripheral sympathetic nerve impulse traffic and cardiac adrenergic drive, and the beneficial effects of ACE inhibitors appear to be especially prominent in patients with increased adrenergic activation. Conversely, β-blockade reduces the secretion of renin, therefore reducing the levels of angiotensin and aldosterone.
Arginine vasopressin is synthesized in the hypothalamus, and its release from the hypophysis is enhanced by osmolar stimuli as well as elevated concentrations of norepinephrine and angiotensin II. Increased release of arginine vasopressin in HF causes vasoconstriction and myocardial fibrosis (through binding to vasopressin 1 receptors) and water retention with dilutional hyponatremia (through binding to vasopressin 2 receptors).
The vasodilator peptides, such as atrial natriuretic peptide (ANP) and β-natriuretic peptide (BNP), are overexpressed in HF (as result of increased wall stress) and lead to a lowering of the right atrial, pulmonary artery, and pulmonary artery wedge pressures. In addition, systemic vascular resistance declines and cardiac output improves, circulating aldosterone and norepinephrine levels decrease, and natriuresis and glomerular filtration increase. However, in many patients with HF, the natriuretic peptides become ineffective despite high circulating levels. The profound activation of the RAAS and adrenergic system may overcome the ability of natriuretic peptides to increase sodium excretion or reduce afterload. In addition, most of the circulating natriuretic peptides in advanced HF patients are secreted as biologically inactive (“defective”) molecules. Finally, changes in renal hemodynamics and a combination of receptor downregulation and increased cyclic guanosine monophosphate phosphodiesterase activity lead to a decreased natriuretic peptide response with consequent increase in salt retention and further deterioration of renal function and increased vasoconstriction.
The development of myocardial hypertrophy, as consequence of neurohormonal activation, initially represents an important adaptive mechanism to hemodynamic stress and is characterized by structural changes at the myocyte level that are translated into alterations in chamber size and geometry. In addition to myocyte alterations, the cardiac fibroblasts and the increased production of extracellular matrix participate in the remodeling process. Increased hemodynamic stress results in changes in myocardial gene expression leading to an impairment of contractile function. In patients with HF, a marked increase in the ventricular end-diastolic pressure may be responsible for changes in the shape of the left ventricle from an ellipsoid to a more spherical configuration. These changes in ventricular geometry result in papillary muscle rearrangement and secondary mitral insufficiency. In addition, the elevated ventricular end-diastolic pressures cause subendocardial ischemia that is perpetuated by compensatory tachycardia, which shortens the diastole and decreases the coronary filling time. This is reflected at a biochemical level by increased production of lactate, adenosine triphosphate, and creatine phosphate and at a histologic level through fibrosis, lowering the threshold for malignant ventricular arrhythmias.
The myocardial fibrosis and chamber dilatation lead to electrical dyssynchrony by fibrosis of the conduction system that, in turn, results in mechanical dyssynchrony (observed in about 40% of the systolic HF patients). When mechanical dyssynchrony is present, contraction and relaxation of the lateral wall occurs earlier than that of the interventricular septum, leading to decreased stroke volume and cardiac output and eventually to secondary mitral regurgitation.
In systolic HF patients, the decreased cardiac output leads to renal hypoperfusion, and the fluid retention leads to increased venous congestion, resulting in further reduction in the renal blood flow and relative renal vasoconstriction, which further exacerbate the sodium and water retention. In addition, the elevated cytokine levels and relative resistance of the bone marrow to erythropoietin lead to anemia in many HF patients. In these patients, peripheral perfusion decreases as a result of a decreased vascular resistance, leading to neurohormonal activation with subsequent reduction in renal blood flow and kidney function, ultimately resulting in expansion of plasma volumes. The increased plasma volume results in increased cardiac work, which leads to left ventricular hypertrophy and ultimately worsens the cardiac function, which in turn worsens renal function, completing the vicious circle (cardio-renal-anemia syndrome).
The chronic elevation in the left ventricular filling pressures that occurs in the setting of left ventricular remodeling and dilatation will lead to an increase in pulmonary venous pressures and a passive increase in pulmonary artery pressures. Due to the chronic elevation in pulmonary arterial pressures (secondary pulmonary hypertension), the pulmonary vascular resistance increases, increasing the right ventricular afterload and leading to deterioration in the right ventricular function over time (right HR). This eventually leads to a decrease in both right and left ventricular stroke volume, which is associated with signs and symptoms of both congestion (dyspnea, peripheral edema, hepatomegaly, elevated jugular venous pressures) and low cardiac output (mental status changes, renal dysfunction, fatigue).
Eventually, the progressive compensatory mechanisms described earlier are overwhelmed and the patients become symptomatic and present to medical attention, often in a hospital setting. In most patients, the condition is progressive, and further deterioration occurs at a myocardial/renal/hepatic/skeletal muscle level, and patients end up having recurrent hospitalizations. There is clear evidence that each hospitalization for HF leads to further myocardial necrosis (evidence by troponin release) and renal injury, which in turn will worsen the overall prognosis and lead to progressive HF and death.
In rare circumstances, a hyperkinetic circulatory state associated with vasodilatation occurs (usually due to an increased demand on the heart from another condition, such as anemia, or thyrotoxicosis) and can trigger HF when superimposed on an underlying heart disease. Fortunately, this is often reversible by prompting treating the associated triggering condition.
Clinical Findings
One of the first principles in the clinical evaluation is to determine whether the patient is indeed presenting with HF, since the presenting signs and symptoms are often nonspecific. Despite a careful history and physical examination, additional diagnostic information may be necessary to support the diagnosis and to determine the mechanism of the symptoms, the severity of the condition, and the prognosis for the individual patient.
The cardinal symptoms of HF are dyspnea and fatigue that occur with exertion and/or at rest, although some patients may present with acute pulmonary edema. Dyspnea is a subjective feeling of air hunger and is the most frequent symptom in HF patients. Initially, dyspnea on exertion will be noted by a change in the extent of physical activity that causes the shortness of breath. As the HF worsens, the intensity of exertion required to produce dyspnea will decrease, and eventually the dyspnea will be present at rest. Interestingly, the severity of dyspnea decreases after right ventricular failure develops.
Paroxysmal nocturnal dyspnea occurs after the patient has been in the supine position for some time. The patient wakes up suddenly with a sensation of choking and air hunger and assumes an upright posture to relieve the symptoms. Paroxysmal nocturnal dyspnea usually precedes orthopnea, may be associated with bronchospasm and wheezing (cardiac asthma), and is caused by a mobilization of interstitial fluid from infradiaphragmatic locations during recumbency with a resultant increase in the circulating blood volume and increased pulmonary venous pressure.
Orthopnea (dyspnea occurring within a few minutes after the patient assumes a supine position) has the same cause as paroxysmal nocturnal dyspnea, but represents a more severe cardiac dysfunction. While elevating the head of the bed, sitting up, or standing can improve the symptoms, the most severely affected patients usually sleep sitting upright.
Another manifestation of pulmonary congestion is a dry or nonproductive cough (especially at night) that is usually relieved by treating the elevated left-sided filling pressures.
As with dyspnea, the mechanism of fatigue in patients with systolic HF is unclear. Although the fatigue was attributed for many years to a low cardiac output, there is mounting evidence of contribution of abnormal skeletal muscle metabolism with shifts in fiber distribution (increase in the percentage of the fast-twitch, glycolytic, easily fatigable fibers) and muscle atrophy, even in the presence of a normal cardiac output. Chronic fatigue begets further inactivity, which leads to further deconditioning and a greater extent of disability.
Other disabling symptoms in HF patients include extreme thirst (associated with activation of the arginine–vasopressin system), nocturia (due to enhanced urine formation in the recumbent position when renal vasoconstriction decreases and venous return to the heart increases), oliguria (usually associated with either increased venous congestion or markedly reduced cardiac output), cerebral symptoms—memory impairment, confusion, insomnia—(usually markers of reduced cardiac output), and ascites and abdominal fullness, accompanied by right upper quadrant pain, early satiety, and nausea (usually related to passive hepatic and splanchnic congestion).
Although most patients with HF appear well nourished, those with more advanced disease (especially in the setting of chronic splanchnic congestion) frequently appear malnourished. This appearance arises from poor caloric intake (due to early satiety and nausea) and impaired absorption of fat and, occasionally, a protein-losing enteropathy, in combination with an increased metabolism due to elevated levels of norepinephrine and tumor necrosis factor-α. The combination of reduced intake and increased energy expenditure leads to a reduction of tissue mass and, in some cases, to cardiac cachexia.
Patients with systolic HF frequently show evidence of increased sympathetic activity such as diaphoresis, pallor and coldness of the limbs, peripheral cyanosis of the digits, abnormal distention of the superficial veins, and compensatory sinus tachycardia (when the cardiac output is decreased).
Cheyne-Stokes respiration (alternating phases of hyperpnea and apnea in a crescendo-decrescendo manner) can be seen in patients with systolic HF and represents an altered neurogenic control of respiration, facilitated by a combination of pulmonary congestion, prolonged circulation time from lung to the brain, and decreased sensitivity of the respiratory center to hypercapnia and hypoxia.
Pulmonary rales can be heard at the lung bases, and they are a result of the transudation of fluid into the alveoli that subsequently moves into the airways. In pulmonary edema, coarse rales and wheezes are heard all over the lung fields and may be accompanied by frothy sputum. With progressive fluid accumulation, pleural effusions (hydrothorax) occur and will increase the severity of dyspnea by further reducing pulmonary vital capacity. Dullness on percussion is the characteristic sign, and shifting dullness can be found when the patient moves from the sitting to the lateral decubitus position. Breath sounds over the area of effusion are diminished or absent. Hydrothorax usually is reabsorbed slowly as diuresis ensues, but in many cases, the pleural effusion lags behind for many days after the symptoms disappear.
Symmetric pitting edema is common in systolic HF patients and involves the dependent portions of the body with higher venous pressure: feet and ankles of ambulatory patients and the sacral area of bedridden ones. With progressive, untreated disease, the edema becomes massive and generalized (anasarca), involving the genital area, the upper extremities, and the thoracic and abdominal walls. Occasionally, in massive overload, skin breakdown and extravasation of fluid can occur. Chronic edema results in increased pigmentation, reddening, and induration of the skin of the lower extremities (stasis dermatitis).
An important part of the volume status assessment is detecting the abnormal distention of the internal jugular veins. Patients should be examined while they are lying down, with the torso and head tilted at 45 degrees. A persistent elevation of the jugular venous pressure is one of the earliest and most reliable signs of HF and is a powerful prognostic indicator of increased risk of HF hospitalizations and all-cause mortality. The inability of the right ventricle to accept transient increases in venous return (positive hepatojugular reflux) is observed during transient compression (30–60 seconds) of the upper abdomen. Passive hepatic congestion is identified by epigastric fullness, hepatomegaly and tenderness on palpation, and dullness to percussion of the right upper quadrant. As with the hydrothorax, these findings may persist after other signs of HF have disappeared because it takes longer for hepatic congestion to disappear. Occasionally, systolic pulsations of the liver may be felt in patients with severe tricuspid regurgitation.
Although rarely used, the hemodynamic response to the Valsalva maneuver is simple to perform and carries one of the best combinations of specificity (91%) and sensitivity (69%) for the detection of elevated left ventricular filling pressures. The maneuver is performed with the blood pressure cuff inflated 15 mm Hg over the systolic blood pressure. While the physician auscultates over the brachial artery, the patient is asked to perform a forced expiratory effort against a closed airway (the Valsalva maneuver). In a normal response, the Korotkoff sounds disappear during sustained maneuver and reappear after release of the maneuver. An abnormal response, in which blood pressure sounds either do not reappear after the maneuver (absent overshoot) or are maintained throughout the maneuver (square wave), is found in patients with systolic HF and elevated left ventricular filling pressures.
Cardiomegaly, with a laterally displaced and sustained point of maximal impulse, can be found on physical examination, but this is a nonspecific finding. The decrease in ventricular compliance becomes apparent by the presence of a late diastolic atrial sound (S4 gallop). A protodiastolic sound (S3 gallop), better heard in more tachycardic patients, is caused by acute deceleration of ventricular inflow after the early filling phase and is a marker of increased left ventricular filling pressures. Together with elevated jugular venous pressure, the presence of a third heart sound is a powerful predictor of outcome and is associated with an increased risk of death and hospitalization for HF. Systolic murmurs are common in HF patients and are secondary to functional mitral or tricuspid regurgitation that results from ventricular dilatation.
Pulsus alternans (a regular rhythm of alternating strong and weak pulsations) is common in patients with systolic HF and is due to an alternation in the stroke volume of the left ventricle. If persistent, it usually implies advanced HF and a chronic low output state.
It is important to emphasize that HF remains a clinical diagnosis. While the symptoms, signs, and clinical findings can, in isolation, be ascribed to other conditions, a systematic integrated approach establishes the diagnosis of HF. Several scoring systems have been used in epidemiologic and clinical studies to diagnose HF, and the Framingham Heart Study criteria have been shown to have the best sensitivity and specificity (Table 26–1). Once the HF is diagnosed, it is important to determine the clinical profile (Figure 26–1) and the severity of symptoms (New York Heart Association functional class), which will dictate the subsequent therapy.
Major Criteria | Minor Criteria |
|---|---|
Paroxysmal nocturnal dyspnea or orthopnea | Dyspnea on exertion |
Elevated jugular venous pressure | Night cough |
Hepatojugular reflux | Tachycardia > 120 bpm |
Pulmonary rales | Hepatomegaly |
S3 gallop | Bilateral lower extremity edema |
Enlarging heart silhouette on consecutive chest x-rays | Pulmonary vascular engorgement on chest x-ray |
Pulmonary edema on chest x-ray | Pleural effusion on chest x-ray |
Weight loss on diuretics (> 10 lb in 5 days) | |
Increased venous pressure > 16 cm H2O (assessed via a central line) |
Figure 26-1
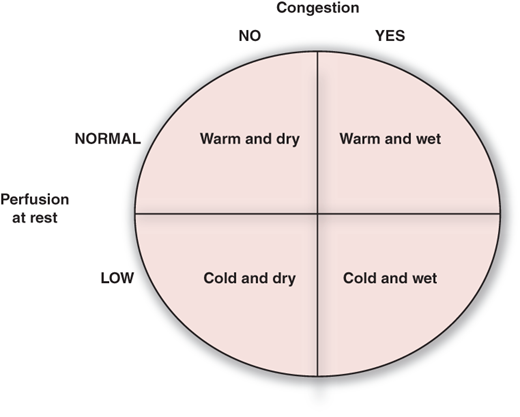
Presentation of heart failure based on assessment of congestion and perfusion. ACE, angiotensin-converting enzyme.
Signs and symptoms of congestion: orthopnea, paroxysmal nocturnal dyspnea, jugular venous distension, positive hepatojugular reflux, pulmonary rales, hepatomegaly, symmetric pedal edema, and Valsalva maneuver square wave.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


