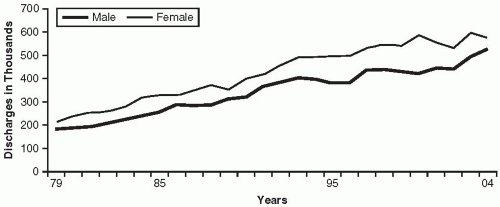
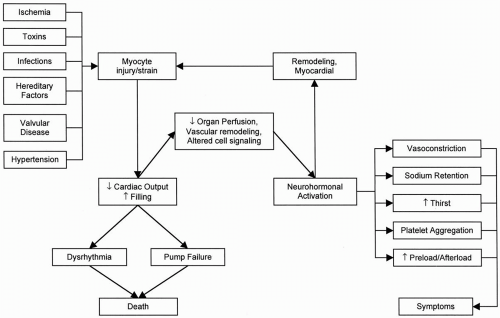
The term
cardiomyopathy is an ill-described, general category for a large group of unrelated disease processes that share only the clinical characteristic of substantially reduced cardiac pumping function and power output. Practical and graphic descriptions have been used to describe cardiomyopathy, and this classification is firmly anchored in the pathologic description of the heart. Thus
dilated cardiomyopathy, hypertrophic cardiomyopathy, and
infiltrative cardiomyopathy are descriptive pathologic terms. Alternatively, cardiomyopathy is characterized by the specific clinical or disease process with which it is associated. Thus terms such as
peripartum cardiomyopathy, diabetic cardiomyopathy, and
toxic cardiomyopathy are used. In clinical practice, the etiology of left ventricular systolic dysfunction can be identified in patients with coronary artery ischemia or infarction, infectious vectors, toxins, hereditary conditions, and conditions for which no cause can be identified (idiopathic) (
Table 14.1). These disorders, in aggregate,
account for the majority of cases of heart failure resulting from myocardial disease.
Ischemia
In patients with coronary artery disease, a cardiomyopathy can develop as the result of one extensive myocardial infarction, multiple smaller myocardial infarctions, ongoing ischemia from severe triple-vessel disease, or coronary artery disease associated with significant mitral regurgitation. Myocardial dysfunction may also develop after coronary artery bypass surgery, even in the setting of otherwise technically adequate graft placement. Early identification of myocardial dysfunction associated with coronary artery disease is important, in view of the potential for reversal of dysfunction with effective management. When overt angina is not apparent, because of limited exercise capacity, subclinical myocardial ischemia may nonetheless produce abnormal systolic and diastolic ventricular dysfunction. Additional subclinical loss of viable myocardium also may occur.
When ischemia-related myocardial dysfunction prevents identification of reversible disease during exercise, viable hibernating
myocardium may be identified by nuclear imaging studies. The predictive value of
201T1 and
99mTc scintigraphy for detecting hibernating myocardium has been enhanced by newer redistribution and reinjection protocols, and these techniques are more readily available than is positron-emission testing at most institutions. In addition, newer techniques using magnetic resonance imaging also are being used for diagnosis of viable myocardium. Once identified, reversible ischemia caused by hibernation can be managed with interventional or surgical techniques, to prevent further deterioration of myocardial function.
Infections
A long list of infectious agents can be identified for this subgroup of myocardial disease. Myocardial dysfunction develops from a nonspecific immune or inflammatory response, or both, or from structural damage to cardiac myocytes. “Viral” myocarditis is frequently suspected among the patients who are otherwise classified as having idiopathic cardiomyopathy, although many of these patients may actually have an inherited cardiomyopathy. Myocarditis can be diagnosed with endomyocardial biopsy and has been shown to be present in 12% of patients seen with dilated cardiomyopathy in the absence of coronary artery disease within 6 months of their original diagnosis (
2). With the exception of a few welldescribed viral causes that can be inferred from serial immune titers, isolation of a specific viral vector remains difficult. Molecular biologic techniques permit enhancement of viral messenger ribonucleic acid. However, in evaluations of myocardial tissue by histologic techniques, the occurrence of viral particles is equally distributed between patients with active myocarditis and those with nonspecific cardiomyopathy. Etiologic origin is therefore difficult to assign.
Diagnosing and treating active myocarditis remains a challenge. In general, the diagnosis can be confirmed only on myocardial biopsy, and the occurrence may be sporadic and related to fluctuation in Coxsackie virus prevalence. Endomyocardial biopsy also can be used for histologic documentation when a strong clinical suspicion of myocarditis is present. Endomyocardial biopsy evidence of myocarditis, on the whole, carries a prognosis similar to that for heart failure of idiopathic origin.
Among patients with history of a flulike syndrome in the setting of clinical suspicion of myocarditis, biopsy evidence of inflammatory infiltrates is seen in approximately 25%, and yet almost half of these patients may have other concurrent disorders. Investigators have shown that the shorter the duration of illness, the greater the likelihood of a biopsy sample positive for inflammation; the likelihood reaches almost 90% in patients seen within 4 weeks of their initial symptoms (
3). Seasonal and yearly variations exist in the clinical presentation. The Myocarditis Treatment Trial demonstrated a better-thanexpected prognosis in patients with myocarditis (
4). Specific immunosuppression did not alter outcome. Additional analyses are in progress to determine immunologic markers that identify patients who may benefit from immunosuppressive therapy.
Myocarditis may be present in cases in which the biopsy result is negative. One possible explanation is that the transvenous technique does not sample enough myocardial sites to detect each case of a disease that may have a focal or multifocal distribution. Acute myocarditis may occur as a regional process and can mimic acute myocardial infarction, with striking electrocardiographic changes. The regional nature of this disorder is evident with invasive and noninvasive assessment of ventricular performance. Depending on clinical presentation, this disorder may necessitate cardiac catheterization, which is the only means of definitively excluding epicardial coronary artery disease.
Myocardial dysfunction due to Chagas disease remains the most common worldwide cause of cardiomyopathy (
5). Infection in humans is caused by a bite from the reduviid insect, which harbors the protozoa
Trypanosoma cruzi in its gastrointestinal tract.
T. cruzi is the infectious etiology of Chagas disease and gains entry into a human host by fecal deposition after a bite
from the reduviid. After initial infection, acute trypanosomiasis occurs, followed by a long latent period; chronic Chagas disease appears up to 20 years later. Myocardial dysfunction and congestive heart failure develop during this time.
T. cruzi parasites within a cellular infiltrate may be present during the acute phase, but cardiac manifestations occur during the chronic phase. No correlation is found between the severity of disease and parasitemia. T lymphocytes may destroy normal myocardial cells, and antibody-mediated responses to specific myocyte components, such as the sarcoplasmic reticulum, have been identified.
In the Western Hemisphere, the greatest concentration of this disorder is in Central and South America, where 20 million people may be infected with the parasite (
5). However, with increasing migration from these regions to the United States, consideration must be given to this diagnosis in patients of Latin American or South American origin from endemic regions. The concern regarding Chagas disease as an etiology of myocardial dysfunction is not limited to the endemic populations. Currently, donor blood products are not routinely screened for
T. cruzi. Thus blood donation is conceivably a means by which
T. cruzi may be spread by a nonvector pathway. An acceptable method to screen blood products routinely for
T. cruzi has not been established. This has not proved to be a significant problem at the current time, but as much as 10 to 15 years may separate acute parasitemia and overt myocardial dysfunction.
Toxins
Several exogenous toxins are well known to cause left ventricular systolic dysfunction and subsequent heart failure. The most common of these is alcohol, represented in 3.4% of cases of systolic heart failure in the absence of coronary artery disease. Myocardial toxicity due to anthracyclines (e.g., doxorubicin) is a common cause of systolic heart failure in patients who received chemotherapy for the treatment of cancer (
6). Other exogenous toxins known to lead to systolic heart failure include cocaine, other chemotherapeutic agents (cyclophosphamide and trastuzumab), interferon, interleukin-2, and chloroquine. In the mid-1960s, a clustering of cases of acute-onset cardiomyopathy developing in patients with heavy beer consumption led to the discovery that cobalt represented a myocardial toxin. When the practice of adding cobalt to beer was stopped, no further cases occurred.
Alcoholic cardiomyopathy
Alcohol may be associated with heart failure in several different ways. Alcohol causes an acute depressant effect on myocardial contractility that can result in measurable dysfunction with binge drinking. Evidence suggests that the fundamental mechanism of injury induced by ethanol is structural and chemical disorganization of membranes, interference with ion transport, and derangement of various biochemical functions that possibly allow calcium to accumulate in the cell (
7). Heavy alcohol consumption may also cause atrial tachyarrhythmias, termed
holiday heart, which may contribute to the development of systolic dysfunction. The amount of alcohol necessary to cause this is unknown, because the testimony of alcoholic patients regarding intake cannot be validated. Studies have shown that ejection fraction correlates inversely with reported alcohol intake in alcoholic patients, and women may be more sensitive to the myocardial toxicity of alcohol than are men.
The pathologic and physiologic characteristics of alcoholic cardiomyopathy are similar to those of idiopathic dilated cardiomyopathy in gross appearance. The morphometric evaluation of endomyocardial biopsy does not provide adjunctive prognostic information in these patients. As many as one fourth of patients with systolic failure due to alcohol may have elevated cardiac output, caused by concomitant liver disease and development of arteriovenous fistulae. Patients with alcoholic cardiomyopathy may have a favorable prognosis in comparison with those with idiopathic cardiomyopathy; approximately 50% of alcoholic patients experience improved
left ventricular function once abstinence is established.
Athracycline and anticancer-related cardiomyopathy
Doxorubicin and daunorubicin are anthracycline analogues that are widely used as chemotherapeutic agents. One important side effect caused by anthracyclines is cardiotoxicity. Cardiotoxicity due to anthracycline derivatives has been shown to be dependent on the cumulative dose (
6). Measurable left ventricular systolic dysfunction is rare in patients receiving less than 350 mg per square meter of doxorubicin but may be seen in as many as 30% of patients receiving more than 600 mg per square meter (
8). The peak levels of the drug may be a determinant for developing the disorder: Some evidence suggests that giving the same total dose weekly rather than every 3 weeks or administering the drug by slow continuous infusion rather than by bolus may reduce the incidence of cardiotoxicity. The risk factors for development of doxorubicin-induced cardiomyopathy include age older than 70, use in combination with other chemotherapeutic agents, concomitant or prior mediastinal radiotherapy, prior cardiac diseases, hypertension, liver disease, and whole-body hyperthermia. Most authors on this topic have recommended monitoring patients serially with radionuclide ventriculography as patients are treated with anthracyclines (
6). The diagnostic test with the greatest specificity and sensitivity for doxorubicin-induced cardiomyopathy is endomyocardial biopsy. Endomyocardial tissue from the right ventricle shows typical histopathologic changes, including loss of myofibrils, distention of the sarcoplasmic reticulum, and vacuolization of the cytoplasm; these may appear before measurable changes in left ventricular systolic function occur. A biopsy scoring system has been described to show that in patients in whom more than 25% of cells exhibit histopathologic changes, substantial changes in the ejection fraction will probably develop, which suggests that treatment should be terminated (
9). However, it is still possible that in patients with lower biopsy grades, cardiomyopathy will develop 4 to 20 years later.
In addition to toxicity related to anthracyclines, several other anticancer therapeutics are associated with development of cardiomyopathy, the most common of which is trastuzumab (Herceptin), used for the treatment of breast cancer. The frequent use of multiple anticancer therapies (including radiation therapy) can greatly increase the future likelihood of developing cardiomyopathy.