34 Guidelines for Managing Pacemaker and Implantable Defibrillator Advisories
Pacemakers and implantable cardiac-defibrillators (ICDs), two of the most remarkable technologic advances in medical history, have undergone significant product modifications since the first human implants.1 For example, although pacemakers became a clinically useful tool in the 1950s, initial models were large, electrically powered, and impractical for long-term patient use. Similarly, early ICDs, initially implanted in humans in 1980 and FDA approved in 1985, were cumbersome, required a thoracotomy for implantation, and had limited reprogramming and data storage capabilities.1,2 Since then, remarkable advances in pacemaker and ICD technology have resulted in substantial reductions in generator size, an exponential increase in computer memory, and advances in lead technology that have reduced diameter, improved chronic pacing thresholds, and with the advent of transvenous leads, greatly facilitated safe lead implantation.3,4 The resultant marked reduction in morbidity and mortality associated with device implantation has led to the widespread adoption of implantable arrhythmia device therapy to the benefit of many patients.
 Pacemaker and ICD System Performance
Pacemaker and ICD System Performance
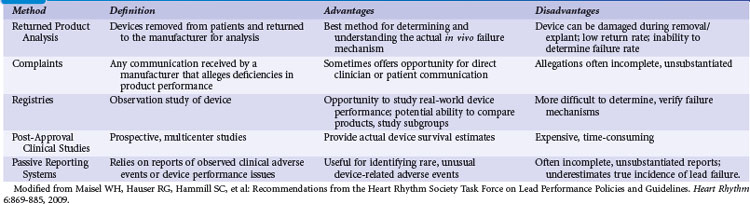
Definitions and Assessment of Device Performance
Pacemaker and ICD systems, consisting of a generator and lead, must be able to reliably monitor cardiac electrical activity and deliver life-sustaining therapy to the heart when needed. Device reliability measures the freedom of an ICD or pacemaker generator or lead from specific structural and electrical failures.5,6 Reliability is usually reported as the percentage of devices still implanted and functioning appropriately at a given point in time (prevalence) or a failure rate per unit of time, such as failure rate per month (incidence). Device performance is a more comprehensive assessment of device quality, clinical usability, freedom from failure (malfunction), and conformance to applicable regulatory labeling. Device performance depends on a number of factors, including device design; materials and manufacturing methods; implanting physician skill and technique; patient characteristics (e.g., age, anatomy, activity level); and the expertise of the caregivers providing postimplant care. Box 34-1 highlights common terms used to describe device performance.5,6
Box 34-1
Definitions of Device Performance Terms
Pacemaker and ICD generators and leads may be removed from service because they are malfunctioning or for reasons unrelated to device performance (e.g., patient death, device infection, device upgrade). A malfunction occurs when an implanted device fails to meet its performance specifications or otherwise perform as intended. Although ideally the mechanism of a device malfunction should be confirmed by direct bench laboratory analysis, in many cases this is not possible. For example, malfunctioning ICD leads often are not explanted because of the hazards associated with lead extraction, or leads are damaged by the extraction process itself, making analysis difficult. In addition, devices may fail to perform as expected in ways not identifiable by bench analysis; for example, a design flaw resulting in an increased risk of perforation or dislodgment may be difficult to distinguish from a complication caused by a physician’s implant technique.6 Accurate and useful assessment of overall device performance and malfunctions demands monitoring, analysis, and reporting not only of device failures, but also of adverse clinical events when such events may be caused by device performance issues.
Generator Performance and Mechanisms of Malfunction
The importance of monitoring pacemaker and ICD performance has long been recognized. For example, the Bilitch Registry was started in 1974 and evaluated 22786 pacemakers from 6 sites over a 19.5-year period and 3520 ICDs from 13 sites over almost 12 years, ending in December 1993.7,8 Failures involving 50 different generator models were reported.7,8 Other long-term device failure registries reported 700 cases of pacemaker and ICD generator malfunction between 1988 and 1996, including 370 “dangerous” cases involving loss of output or decrease in pulse amplitude.7,9
More modern device performance is reflected by data compiled from manufacturer annual reports, submitted to the U.S Food And Drug Administration (FDA) from 1990 and 2002.10 Because pacemakers and ICDs represent “life-sustaining” therapies for many patients, the FDA requires manufacturers to submit annual reports detailing the number of device implants by model number and information concerning device malfunctions. From 1990 to 2002, 17,323 devices (8834 pacemakers and 8489 ICDs) were explanted because of confirmed malfunction.10 The annual number of malfunctions ranged from 366 to 1152 for pacemakers and 134 to 2228 for ICDs (Fig. 34-1). The device malfunction replacement rate (the number of devices removed in a given year because of malfunction indexed to the number of device implants in that same year) during the study period was 20.7 per 1000 implants for ICDs and 4.6 per 1000 implants for pacemakers.10
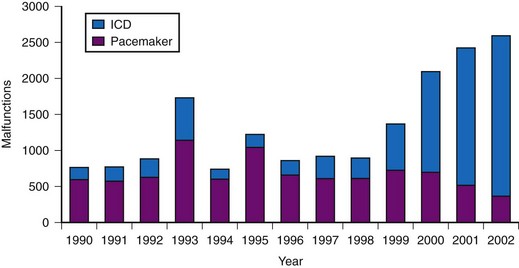
Figure 34-1 Annual number of malfunctions for pacemakers and ICDs (1990-2002).
(Adapted from Maisel WH et al: Pacemaker and ICD generator malfunctions: analysis of Food and Drug Administration annual reports. JAMA 295:1901-1906, 2006.)
A meta-analysis of active device registries (including the Bilitch Registry, the Danish Pacemaker and ICD Register, and the United Kingdom [UK] Pacemaker and ICD Registry) provides additional insights into overall pacemaker and ICD generator performance as well as trends in device reliability.11 During 2.1 million pacemaker person-years of observation over 22 years, pacemaker malfunctions were observed in 2981 devices. Pacemaker reliability improved greatly during the study period, from an annual pacemaker malfunction rate of 12.4 malfunctions per 1000 person-years in 1983 to less than 1.0 malfunction per 1000 person-years in the latter study years (Fig. 34-2). Malfunctions in ICDs were observed during the 17-year study period in 384 devices during 14,821 person-years of observation. Annual ICD malfunction rates ranged from a high of 52.5 malfunctions per 1000 person-years in 1989 to a low of 5.6 malfunctions per 1000 person-years in 1998. A significant improvement in ICD reliability occurred during the decade beginning in 1988 until a transient increase in the malfunction rate was observed in the late 1990s and early 2000s (Fig. 34-3). Device reliability again improved greatly beginning in 2003. Overall, the annual ICD malfunction rate was about 20-fold higher than the pacemaker malfunction rate (26.5 vs. 1.3 malfunctions per 1000 person-years).11
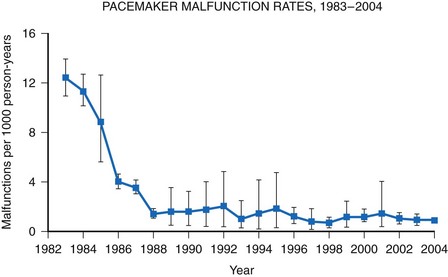
Figure 34-2 Pacemaker malfunction rates: 1983-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
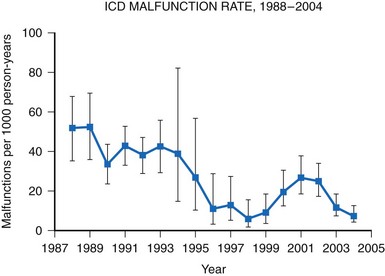
Figure 34-3 Annual ICD malfunction rates: 1988-2004.
(Adapted from Maisel WH: Pacemaker and ICD generator reliability: meta-analysis of device registries. JAMA 295:1929-1934, 2006.)
Analysis of FDA annual reports that include manufacturer-confirmed analysis of device malfunctions demonstrates that hardware abnormalities are by far the most common type of device malfunction requiring device replacement for both pacemakers and ICDs.10 Overall, hardware problems represent 79.8% of observed malfunctions. Of these, battery/capacitor abnormalities (23.6%) and electrical issues (27.1%) accounted for half the total device failures.10 Firmware (integral device software) abnormalities and miscellaneous problems (e.g., physical damage, foreign material contamination, manufacturing errors) were much less frequent causes of device malfunctions. In only 4.7% of cases could the manufacturer confirm device malfunction but not determine the cause. Battery/capacitor abnormalities account for a higher percentage of device malfunctions in ICDs than in pacemakers (31.7% vs. 15.8%).10 Miscellaneous electrical problems, such as broken wires, current leakage, and electrical short circuits, represented similar percentages of malfunctions in pacemakers and in ICDs.
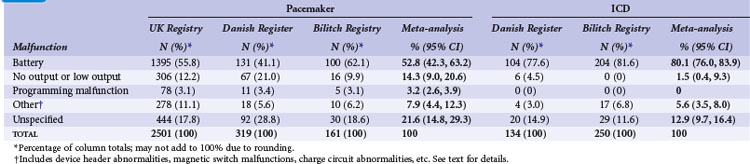
Meta-analysis of active device registries has yielded similar findings.11 Battery malfunctions, most often premature battery failure, were the most common cause of device failure in the Bilitch Registry, the Danish Pacemaker and ICD Register, and the UK Pacemaker and ICD Registry11 (Table 34-1). For pacemakers, an additional 14.3% of device malfunctions were caused by low-output or no-output states. A number of root causes, however, can masquerade as a battery abnormality, including rapid premature battery depletion, electrical short circuits, inappropriate high current drains, or hermetic seal abnormalities. Miscellaneous other types of device failures, such as programming malfunctions, device header abnormalities, magnetic switch malfunctions, and charge circuit abnormalities (for ICDs), accounted for the majority of the other reported device malfunctions.11
Lead Performance and Mechanisms of Malfunction
Pacemaker and ICD lead performance is more difficult to assess than generator performance because malfunctioning leads are often not explanted, and even when they are, leads are often damaged during the explant process.6,12 In addition, reported lead performance varies widely depending on study design, performance definitions, physician and patient characteristics, implant methodology, duration and method of follow-up, and lead models studied. These factors explain some of the observed variability in lead performance.12
Manufacturer product reports describe the performance of atrial, right ventricular, left ventricular, and high-voltage leads and demonstrate lead survival probabilities for most leads of 92% to 99% at 5 years after initial implant.13–17 However, most of these lead survival estimates are significantly limited due to potential underreporting of device malfunctions, insufficient patient follow-up, lead surveillance based on voluntary reporting, and lack of uniform definitions of lead performance and malfunction.6,12 Some manufacturers have utilized prospective, multicenter lead studies in an effort to overcome these limitations. However, small sample sizes or slow enrollment can undermine the ability of these studies to accurately identify underperforming leads in a timely manner.18 Also, they may fail to identify important differences in lead performance between models.
Other data sources also provide estimates of lead reliability. In Denmark, all lead implantations are entered into a longitudinal registry.19 Ten-year lead survival for unipolar and bipolar pacemaker leads implanted since 1993 was reported as 96.5% and 97.8%, respectively, and reliability has improved over time.19
Eckstein et al.20 conducted a retrospective analysis of 1317 consecutive patients who received ICD systems (including 38 different ICD lead models) at three centers in Germany between 1993 and 2004. Follow-up after implantation included noninvasive routine lead evaluation every 3 to 6 months. For the study, “lead failure” was defined as a lead-related problem requiring surgical revision performed at the discretion of the treating physician. Abnormalities were classified as either structural (insulation defects or lead fracture) or functional (far-field sensing; T-wave or physiologic oversensing, noise from contact with another lead, unstable impedance measurements, R-wave reduction, or loss of capture). During a median follow-up of 6.4 years, 38 ICD leads required surgical revision, resulting in a reported cumulative ICD lead survival rate of 97.5% at 5 years.20 A number of prior published studies report on the reliability and durability of ICD leads.21 Reported ICD lead “survival” varies from 91% to 99% at 2 years, 85% to 98% at 5 years, and 60% to 72% at 8 years12,21–33 (Fig. 34-4).
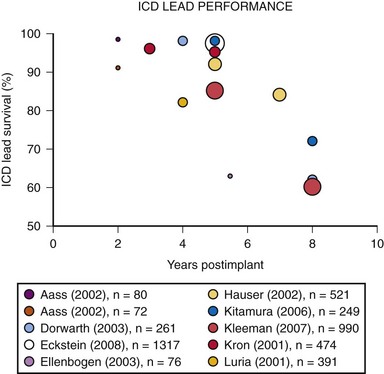
Figure 34-4 Results of selected studies of ICD lead performance.
(From Maisel WH, Kramer DB: ICD lead performance. Circulation 117:2721-2723, 2008.)
Importantly, the definition of ICD lead “survival” or lead “performance” varies among published studies, making it difficult to compare manufacturers or lead models.12 Typically, lead malfunction is defined as electrical abnormalities on lead testing, a chest radiograph consistent with a fracture, or evidence of oversensing unrelated to cardiac signals. Other studies, however, rely on physician clinical judgment and require replacement of the ICD lead in order to consider the lead to have malfunctioned.20 In most published studies, thresholds for action are poorly defined and ambiguous.
Although conceptually simple, pacemaker and ICD leads are complicated devices with lead designs that vary from model to model. These design differences may include variations in insulation, cable/conductor, length, diameter, and fixation mechanism.34 Knowledge about lead abnormalities mainly comes from manufacturer analysis of returned products, which is a rich source of information that manufacturers utilize to support product improvements and enhancements.4 Mechanical or electrical abnormalities may develop in components such as insulation, conductors, the connector, the terminal pin, or the stimulation electrode. In addition to failure mechanisms that are intrinsic to the lead, extrinsic factors (lead damage from trauma, mishandling, lead dislodgment, etc.) may cause pacemaker and ICD leads to provide insufficient therapy.6
The clinical implications of a lead malfunction vary depending on the type of malfunction and the individual patient’s clinical condition. Notably, some structurally normal leads may provide insufficient therapy (e.g., lack of clinical improvement with cardiac resynchronization therapy), inappropriate therapy (e.g., shock delivery for rapid atrial fibrillation), or may need to be removed from service as a result of issues unrelated to the lead, such as the patient’s underlying illness, physiology, implant technique, or device upgrade (e.g., from pacemaker to ICD).6 Importantly, patient and physician characteristics—in addition to lead design—affect ICD lead performance.4 For example, patients with an ICD lead failure are eight times more likely to experience a second failure than a patient without a failure.20
Unfortunately, the tools available to detect impending ICD lead failure are limited. Fluoroscopy, radiography, electrical testing, or direct visualization may be used to detect lead abnormalities, but in many respects these methods are quite rudimentary, imprecise, and insensitive. Long-term, remote monitoring has provided insights into the sometimes intermittent nature of lead abnormalities and has allowed earlier detection of lead performance issues.12
The Heart Rhythm Society (HRS) Task Force on Lead Performance Policies and Guidelines proposed a classification scheme for pacemaker and ICD leads removed from service6 (Fig. 34-5).

Surveillance of Device Performance
The goal of post-market surveillance is to “enhance the public health by reducing the incidence of medical device adverse experiences.”35 The current surveillance system relies on regulators, medical device manufacturers, health care providers, hospitals and other medical care facilities, and patients to report device malfunctions.
Regulatory authorities utilize several different methods to conduct post-market surveillance. For example, the FDA uses spontaneous reporting systems, analysis of large health care databases, scientific studies, registries, and field inspection of facilities5,6,35 (Table 34-2). Historically, the FDA has depended primarily on a passive, “adverse event” reporting system, relying on patients and the health care industry to identify and report adverse events, including rare but serious occurrences. Manufacturers are required to report to the FDA any medical device–related event or malfunction that may have caused or could cause a serious injury or death. Hospitals, nursing homes, and other medical facilities are required to report serious device-related injuries to the manufacturer and device-related deaths to both the manufacturer and the FDA.
The FDA annually receives more than 220,000 adverse event reports regarding medical devices of all types, including some that involve pacemakers, ICDs, or leads.36 The vast majority of reports are provided by manufacturers; fewer than 10,000 come directly from medical facilities. Postmortem device interrogation is rarely performed.37 Health care professionals and patients are encouraged, but not required, to report suspected device-related adverse events, but the FDA receives only several thousand reports from health care providers annually, and physicians, in particular, rarely report events.4–6,35,36
The Manufacturer and User Facility Device Experience (MAUDE) database was established to assist with adverse event reporting and information dissemination for medical devices of all types.38 It contains more than 1 million adverse event reports, including voluntary reports since June 1993 and manufacturer reports since August 1996.35,38 Selected information from this database is publicly searchable via the Internet, and the database has been successfully used to identify concerning safety signals.39,40 However, because submitted adverse event reports are sometimes cryptic or incomplete, it is often difficult to determine if a true device malfunction or patient injury has occurred. Furthermore, significant underreporting of device malfunctions and the absence of denominator data make it difficult to determine the true rate of an observed abnormality41 (Fig. 34-6). For example, all product experience reports from Italy related to one ICD lead were reviewed and demonstrated that only 126 of 454 physician product experience reports were accompanied by a returned lead—and 80% of the returns were related to implant procedures, not chronic use.42
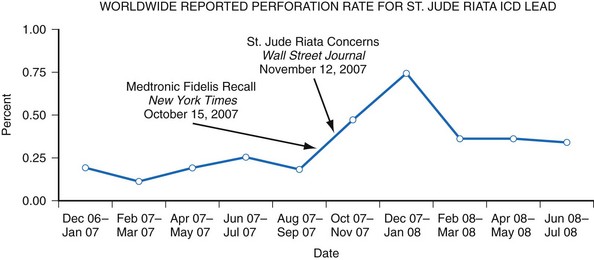
Figure 34-6 Worldwide St. Jude Riata ICD lead reported perforation rate.
(From Maisel WH: Implantable cardioverter-defibrillator lead complication: when is an outbreak out-of-bounds? Heart Rhythm 5:1673-1674, 2008.)
Recognizing this shortcoming, the FDA established the Medical Product Safety Network (MedSun) and HeartNet.43 This active surveillance system utilizes health care facilities and individuals specially trained in device adverse event reporting to identify problems in both device function and user error in the clinical setting. HeartNet in particular focuses on identifying, understanding, and solving problems with medical devices used in electrophysiology laboratories.43 In addition, many manufacturers utilize post-market studies to track the performance of their products. The Centers for Medicare and Medicaid Services (CMS) mandated National Cardiovascular Data Registry (NCDR) collects data on ICD generators and leads and is being better adapted to track longitudinal device performance.44,45 Remote monitoring of device performance has revolutionized pacemaker and ICD patient care by providing automated checks of device functionality and wireless notification of device performance to manufacturers and health care providers46 (see Chapter 32).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


