to uncover interchromosomal rearrangements and have been successful in revealing subtle karyotypic alterations, which would be otherwise overlooked.
 FIGURE 6.1 SKY of a primary lung adenocarcinoma showing numerous numerical and structural chromosome changes. The classifiied image with the pseudocolors is shown in (A) and the inverted-DAPI image is shown in (B). The specimen was near triploid, with extra copies of chromosomes 1, 10, 12, 19, and 20 and deletions of segments of chromosomes 1, 4, 5, 6, and 11. Translocations were found involving chromosomes 1 and 13, 1 and 17, 3 and 20, 5 and 19, 5 and X, 6 and 22, 7, 15, and X, 18 and 21, 16 and 22, and 18 and 21. Two very small marker chromosomes were found carrying chromosome 2 sequences. (See color plate.) |
has started long ago. The first recurrent changes in lung cancer, the deletions of 3p in SCLC, were identified by classical karyotypic analysis.18 However, probably because of the complexity of the chromosomal alterations and the limitations of the conventional techniques, few karyotype reports of primary lung tumors or cell lines were published in the 2 decades following that seminal publication.19,20,21,22,23,24,25,26 Loss of large chromosomal segments in 3p and 8p, gain of whole chromosomal arms, such as 5p, and amplification by homogeneously staining regions (HSR) and double minutes (DM) were reported. However, the conclusions of all those studies were by and large limited, often yielding incomplete karyotypes.
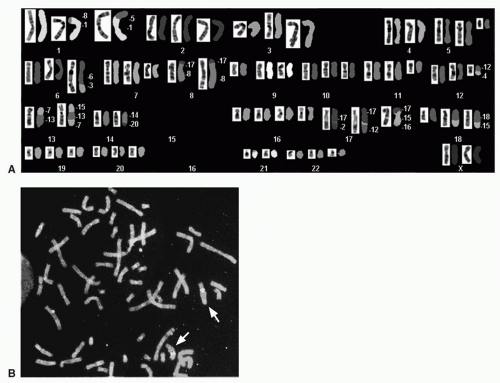 FIGURE 6.2 A: SKY of the non-small cell lung cancer cell line Calu 3 showing multiple abnormalities and two copies of abnormal chromosome 17, derivatives from the translocations between chromosomes 17 and 2 and chromosomes 17 and 12. In addition, the chromosome 17 material identified by SKY in the long arm (q-arm) of these derivative chromosomes was larger than expected if that arm was normal. B: FISH analyses with a probe set including ERBB2 and CEP 17 sequences demonstrated that there was ERBB2 gene amplification in both derivatives (indicated by the arrows). In the FISH assays, ERBB2 probe is highlighted by red color and CEP 17 by green color. (See color plate.) |
Notch3 expression was detected in approximately 40% of resected lung tumors and positively correlated with epidermal growth factor receptor (EGFR) expression. Notch inhibition was shown to increase sensitivity to EGFR tyrosine kinase inhibitors (TKIs) and decrease mitogen-activated protein kinase (MAPK) phosphorylation, observations that support a role for NOTCH3 signaling in lung cancer through EGFR-related pathways.46 The translocation breakpoints were later refined to 15q13.2 and 19p13.1 and the cloning of these regions identified a novel fusion transcript in which the 3′ end of the BRD4 gene on chromosome 19p was fused to the 5′ end of the NUT gene on chromosome 15q. The BRD4-NUT fusion was demonstrated to alter the cell cycle kinetics, augmenting the inhibition of the progression G1 to S phase compared with the wild type BRD4 gene. However, the exact role of the BRD4- NUT fusion in the pathogenesis of lung cancers remains unclear and the t(15;19) has not been found in large lung cancer cohorts tested, which suggest that it is not common in lung cancer.47 Gene fusions detected using other strategies are going to be discussed later. The detection of the intracellular targets of these fusions is expected to bring new insights into molecular pathways that trigger tumor development.
TABLE 6.1 Genomic Regions Showing Gains and Losses in Small Cell Lung Carcinoma Primary Tumors and Cell Lines Detected by Comparative Genomic Hybridization | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The tumor suppressor genes, also known as recessive oncogenes, are inactivated by genetic mechanisms such as point mutations, chromosomal rearrangements, and mitotic recombinations, and by epigenetic events like hypomethylation or hypermethylation of gene promoter regions. It is largely accepted that the inactivation of tumor suppressor genes commonly occurs through a combination of two or more events, the Knudson hypothesis. Still, it is also recognized that the phenomenon in carcinomas is more complex because of mutational instability and chromosomal instability.63 The major tumor suppressor genes involved in lung cancer are TP53 (17p13.1), RB1 (13q14.11), CDKN2 (p16INK4a or MST1, 9p21), and several genes located at the short arm of chromosome 3. The incidence of abnormalities in each of these genes in lung cancer, their main role in the development of the disease, and their contribution as prognostic or predictive markers will be briefly summarized.
TABLE 6.2 Genomic Regions Showing Gains and Losses in Non-Small Cell Lung Carcinoma Primary Tumors and Cell Lines Detected by Comparative Genomic Hybridization | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 6.3 Genomic Regions Exhibiting Focal Amplification in Lung Cancers, Detected by at Least Two Independent Studies Using Comparative Genomic Hybridization Analyses | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
and premalignant lung lesions and restoration of its function in 3p21.3-deficient NSCLC cells significantly inhibits tumor cell growth by induction of apoptosis and alteration of cell cycle kinetics.113 Both SEMA3F and SEMA3B transcripts are underrepresented in lung cancers, mainly squamous cell carcinomas. A recent review114 indicated that downregulation of SEMA3B and SEMA3F is sustained by gene hypermethylation in lung cancer cell lines.115,116 Moreover, the loss of function of these genes correlates inversely with grade and stage of lung cancer.98,117 Additionally, the SEMA3B and SEMA3F genes were found to be targets of TP53,118,119 which suggests that they could be activated during DNA damage or other stress responses. Deregulation in MLH1, a mismatch repair gene, was detected in up to 78% of NSCLC specimens, predominantly
by promoter hypermethylation120 and more recently has been associated with poor prognosis in NSCLC.121 RARB mediates growth control responses122,123 and its expression was found reduced in about 50% of NSCLC and 70% of SCLC.124 Promoter hypermethylation is the leading cause of silencing of this gene. Conclusions have been controversial regarding the prognostic role of RARB suppression in lung cancer125,126 as well as regarding the efficacy of retinoids as chemopreventive agents for this disease.127,128
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


