David J. Tester, Michael J. Ackerman
Genetics of Cardiac Arrhythmias
Cardiac arrhythmias encompass a large and heterogeneous group of electrical abnormalities of the heart, with or without underlying structural heart disease. Cardiac arrhythmias can be innocuous, can predispose to the development of potentially lethal stroke or embolism, or can be an emergency, life-threatening condition that may result in sudden cardiac death (SCD), one of the most common causes of death in the developed countries. In the United States, for example, an estimated 300,000 to 400,000 individuals die suddenly each year, with the vast majority involving the elderly; 80% are caused by ventricular fibrillation in the context of ischemic heart disease. In comparison, SCD in the young is relatively uncommon, with an incidence between 1.3 and 8.5 per 100,000 patient-years.1 However, tragically, thousands of otherwise healthy individuals younger than 40 years die suddenly each year without warning. Most SCD in the young can be attributed to structural cardiovascular anomalies identifiable at autopsy, but in as many as 30% to 50% of such individuals, sudden death remains unexplained following a complete autopsy and medicolegal investigation (see Chapter 39).
Potentially lethal and inheritable arrhythmia syndromes included under the umbrella of “cardiac channelopathies,” such as congenital long-QT syndrome (LQTS), Brugada syndrome (BrS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and related disorders, involve electrical disturbances with the propensity to produce fatal arrhythmias in the setting of a structurally normal heart. These often unassuming electrical abnormalities have the capacity to cause a potentially lethal arrhythmia to develop in the heart of an unsuspecting, otherwise healthy individual and lead to sudden and early demise.1 In fact, it is now recognized that almost a third of autopsy-negative sudden unexplained deaths (SUDs) in the young2 and approximately 10% of cases of sudden infant death syndrome (SIDS) stem from these genetically inherited cardiac channelopathies.3,4
Through molecular advances in the field of cardiovascular genetics, the underlying genetic bases responsible for many inherited cardiac arrhythmia syndromes have come to light, and the underlying genetic substrates responsible for other such syndromes are on the cusp of discovery. Over the past decade, a particular set of themes, including extreme genetic heterogeneity, reduced or incomplete penetrance, and variable expressivity, has proved to be commonplace among the cardiac channelopathies. However, for some disorders, important genotype-phenotype correlates have been recognized and have provided diagnostic, prognostic, and therapeutic impact. The clinical description, genetic basis, and genotype-phenotype correlates associated with these inherited arrhythmia syndromes are discussed in this chapter.
The QT-Opathies
Long-QT Syndrome
Clinical Description and Manifestations of Long-QT Syndrome
Congenital LQTS comprises a distinct group of cardiac channelopathies characterized by delayed repolarization of the myocardium, QT prolongation (QTc >480 msec as the 50th percentile in genetically confirmed LQTS cohorts), and increased risk for syncope, seizures, and SCD in the setting of a structurally normal heart and otherwise healthy individual. The incidence of LQTS may exceed 1 in 2500 persons.5 Individuals with LQTS may or may not manifest QT prolongation on a resting 12-lead surface electrocardiogram (ECG). This repolarization abnormality is almost always without consequence; however, triggers such as exertion, swimming, emotion, auditory stimuli (e.g., alarm clock), and the postpartum period can rarely cause the heart to become electrically unstable and result in the development of the potentially life-threatening and sometimes lethal arrhythmia of torsades de pointes (TdP) (see Chapter 37). Although the cardiac rhythm most often returns to normal spontaneously, with only a transient episode of syncope, 5% of individuals with untreated and unsuspected LQTS succumb to a fatal arrhythmia as their sentinel event. However, it is estimated that nearly half of the individuals experiencing SCD stemming from this very treatable arrhythmogenic disorder may have previously exhibited warning signs (i.e., exertional syncope, family history of premature sudden death) that went unrecognized.2 LQTS may explain approximately 20% of autopsy-negative SUDs in the young and 10% of cases of SIDS.2,3
Genetic Basis for Long-QT Syndrome.
LQTS is a genetically heterogeneous disorder largely inherited in an autosomal dominant pattern; it was previously known as Romano-Ward syndrome. Rarely, LQTS is inherited as a recessive trait first described by Jervell and Lange-Nielsen and is characterized by a severe cardiac phenotype and sensorineural hearing loss. Spontaneous/sporadic germline mutations can account for nearly 5% to 10% of cases of LQTS. To date, hundreds of mutations have been identified in 10 LQTS susceptibility genes responsible for a nonsyndromic “classic” LQTS phenotype. In addition, two extremely rare, multisystem disorders associated with marked QT prolongation: Timothy syndrome (TS), formerly annotated as LQT8, and prolonged QU intervals (Anderson-Tawil syndrome [ATS], formerly annotated as LQT7), and a third disorder, LQT4, which is better classified as ankyrin-B syndrome, have also been described.
Approximately 75% of patients with a clinically robust diagnosis of LQTS host either loss-of-function or gain-of-function mutations in one of three major LQTS genes (Table 32-1)—the KCNQ1-encoded IKs (Kv7.1) potassium channel (LQT1, ≈35%; loss of function), the KCNH2- encoded IKr (Kv11.1) potassium channel (LQT2, ≈30%; loss of function), and the SCN5A-encoded INa (Nav1.5) sodium channel (LQT3, ≈10%, gain of function)—that are responsible for orchestration of the cardiac action potential6,7 (Fig. 32-1). Approximately 5% to 10% of patients have multiple mutations in these genes, and those with multiple LQTS mutations are affected at a younger age and exhibit greater expressivity6 (see Chapter 8). The most recent discovery was reported in 2012 by Boczek and colleagues following whole exome sequencing, genomic triangulation, and a systems biology approach to identify a novel genetic substrate (P857R-CACNA1C) for a large 15-member (8 affected) multigenerational pedigree with autosomal dominant “classic” LQTS.8 Functional characterization of the mutation via a whole-cell patch clamp technique revealed a gain-of-function mutation in peak ICa,L consistent with prolongation of the cardiac action potential and the clinical phenotype of LQTS. Their subsequent mutational analysis of 102 unrelated patients with robust clinical evidence of LQTS has indicated that 3% to 5% of genetically elusive cases of LQTS may be attributed to CACNA1C mutations, thus making CACNA1C potentially the fifth most common genetic substrate for nonsyndromic LQTS. The majority of the mutations identified reside in the CACNA1C-encoded critical PEST domain of the L-type calcium channel (LTCC), which signals for rapid protein degradation. These mutations presumably result in a biogenic increase in LTCCs at the cell surface membrane.
TABLE 32-1
Summary of Heritable Arrhythmia Syndrome Susceptibility Genes
| GENE | LOCUS | PROTEIN |
| Long-QT Syndrome | ||
| Major LQTS Genes | ||
| KCNQ1 (LQT1) | 11p15.5 | IKs potassium channel alpha subunit (KVLQT1, Kv7.1) |
| KCNH2 (LQT2) | 7q35-36 | IKr potassium channel alpha subunit (HERG, Kv11.1) |
| SCN5A (LQT3) | 3p21-p24 | Cardiac sodium channel alpha subunit (Nav1.5) |
| Minor LQTS Genes (Listed Alphabetically) | ||
| AKAP9 | 7q21-q22 | Yotiao |
| CACNA1C | 12p13.3 | Voltage gated L-type calcium channel (Cav1.2) |
| CAV3 | 3p25 | Caveolin-3 |
| KCNE1 | 21q22.1 | Potassium channel beta subunit (MinK) |
| KCNE2 | 21q22.1 | Potassium channel beta subunit (MiRP1) |
| KCNJ5 | 11q24.3 | Kir3.4 subunit of IKACH channel |
| SCN4B | 11q23.3 | Sodium channel beta 4 subunit |
| SNTA1 | 20q11.2 | Syntrophin-alpha 1 |
| Andersen-Tawil Syndrome | ||
| KCNJ2 (ATS1) | 17q23 | IK1 potassium channel (Kir2.1) |
| Timothy Syndrome | ||
| CACNA1C | 12p13.3 | Voltage gated L-type calcium channel (Cav1.2) |
| Short-QT Syndrome | ||
| KCNH2 (SQT1) | 7q35-36 | IKr potassium channel alpha subunit (HERG, Kv11.1) |
| KCNQ1(SQT2) | 11p15.5 | IKs potassium channel alpha subunit (KVLQT1, Kv7.1) |
| KCNJ2 (SQT3) | 17q23 | IK1 potassium channel (Kir2.1) |
| CACNA1C (SQT4) | 12p13.3 | Voltage gated L-type calcium channel (Cav1.2) |
| CACNB2 (SQT5) | 10p12 | Voltage gated L-type calcium channel beta 2 subunit |
| CACN2D1 (SQT6) | 7q21-q22 | Voltage gated L-type calcium channel 2 delta 1 subunit |
| Catecholaminergic Polymorphic Ventricular Tachycardia | ||
| RYR2 (CPVT1) | 1q42.1-q43 | Ryanodine receptor 2 |
| CASQ2 (CPVT2) | 1p13.3 | Calsequestrin 2 |
| KCNJ2 (CPVT3) | 17q23 | IK1 potassium channel (Kir2.1) |
| CALM1 | 14q32.11 | Calmodulin 1 |
| TRDN | 6q22.31 | Triadin |
| Brugada Syndrome | ||
| SCN5A (BrS1) | 3p21-p24 | Cardiac sodium channel alpha subunit (Nav1.5) |
| Minor BrS Genes (listed alphabetically) | ||
| GPD1L | 3p22.3 | Glycerol-3-phosphate dehydrogenase 1–like |
| CACNA1C | 12p13.3 | Voltage gated L-type calcium channel (Cav1.2) |
| CACNA2D1 | 7q21-q22 | Voltage gated L-type calcium channel 2 delta 1 subunit |
| CACNB2 | 10p12 | Voltage gated L-type calcium channel beta 2 subunit |
| DLG1 | 3q29 | Synapse-associated protein 97 |
| KCND3 | 1p13.2 | Voltage-gated potassium channel (Ito) subunit Kv4.3 |
| KCNE3 | 11q13.4 | Potassium channel beta subunit 3 (MiRP2) |
| KCNE5 | Xq22.3 | Potassium channel beta subunit 5 |
| KCNJ8 | 12p12.1 | Inward rectifier K+ channel Kir6.1 |
| HCN4 | 15q24.1 | Hyperpolarization-activated cyclic nucleotide-gated channel 4 |
| MOG1 | 17p13.1 | RAN guanine nucleotide release factor 1 |
| SCN1B | 19q13 | Sodium channel beta 1 |
| SCN3B | 11q24.1 | Sodium channel beta 3 |
| SLMAP | 3p14.3 | Sarcolemma associated protein |
| Early Repolarization Syndrome | ||
| CACNA1C | 12p13.3 | Voltage gated L-type calcium channel (Cav1.2) |
| CACNA2D1 | 7q21-q22 | Voltage gated L-type calcium channel 2 delta 1 subunit |
| CACNB2 | 10p12 | Voltage gated L-type calcium channel beta 2 subunit |
| KCNJ8 | 12p12.1 | Inward rectifier K+ channel Kir6.1 |
| Progressive Cardiac Conduction Disease | ||
| SCN5A | 3p21-p24 | Cardiac sodium channel alpha subunit (Nav1.5) |
| TRPM4 | 19q13.33 | Transient receptor potential cation channel, subfamily M, member 4 |
| Sick Sinus Syndrome | ||
| ANKB | 4q25-q27 | Ankyrin-B |
| HCN4 | 15q24-q25 | Hyperpolarization-activated cyclic nucleotide–gated channel 4 |
| SCN5A | 3p21-p24 | Cardiac sodium channel alpha subunit (Nav1.5) |
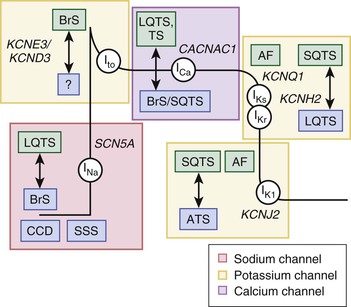
The remaining seven minor LQTS susceptibility genes encode for either cardiac ion channels or key cardiac channel interacting proteins (“ChIPs”) that generally regulate the native ion channel current and collectively explain perhaps 5% of cases of LQTS. The vast majority of LQTS susceptibility mutations consist of single nucleotide substitutions or small insertions/deletions resulting in nonsynonymous missense (amino acid substitution for another amino acid), nonsense (amino acid substitution for a termination codon), or splice site alterations (resulting in exon skipping or intron inclusion) or frameshift mutations (altered normal amino acid coding resulting in early termination).6,7,9 Recently, a few large gene rearrangements involving hundreds to thousands of nucleotides resulting in single or multiple deletions/duplications of whole exons have been described.10,11 Importantly, quintessential mutational “hot spots” are not present within these genes, with the vast majority of unrelated families having their own unique “private” mutation. In 2013 it is important to note that nearly 20% of clinically definite cases of LQTS remain genetically elusive.
In contrast to the rare, pathogenic LQTS-associated channel mutations present in less than 0.04% (1/2500) of persons and in 75% of clinically robust LQTS cases, comprehensive genetic testing for KCNQ1, KCNH2, and SCN5A in more than 1300 ostensibly healthy volunteers has revealed that approximately 4% of white individuals and up to 8% of nonwhite individuals host rare nonsynonymous genetic variants (<0.5% allelic frequency) of these specific cardiac channel genes.12 In fact, a total of 79 distinct channel variants were detected in these healthy subjects, including 14 variants in KCNQ1, 28 in KCNH2 and 37 in SCN5A.12 This has enabled a case-control mutational analysis of the properties and localization of case-associated mutations relative to the compendium of presumably innocuous variants.12 The probabilistic rather than the binary nature of genetic testing is depicted in Figure 32-2, which shows that rare mutations other than missense mutations (approximately 20% of the LQTS spectrum of mutations) are high-probability LQTS-associated mutations whereas the probability of pathogenicity for the most common mutation type, missense mutations (i.e., single amino acid substitutions), is strongly location dependent. For example, missense mutations localizing to the transmembrane-spanning/pore domains of the LQT1- and LQT2-associated potassium channels are high-probability disease mutations, whereas a similarly rare missense mutation that localizes to the domain I-II linker of the Nav1.5 sodium channel is indeterminate, a variant of uncertain significance (VUS). Without cosegregation or functional data, such a mutation has a point estimate for probability of pathogenicity of less than 50%.
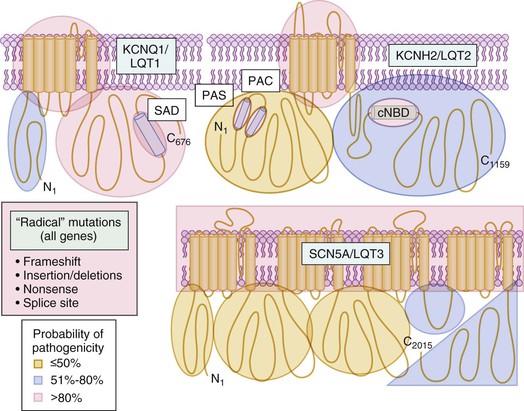
In addition to this background frequency (4% to 8%) of rare variants in health, 15 unique common polymorphisms (allelic frequency >0.5%) have been identified in the four potassium channel subunit genes (KCNQ1, KCNH2, KCNE1, and KCNE2), and 8 common polymorphisms have been identified in the sodium channel gene (SCN5A). Many of these rare and common polymorphisms represent innocent bystanders; however, a layer of complexity is added to the genetics of these channelopathies, and management of patients with otherwise apparently innocuous variants can modify disease. For example, the most common sodium channel variant, H558R, which has a minor allelic frequency of approximately 29% in blacks, 23% in Hispanics, 20% in whites, and 9% in Asians, can provide a modifying effect on the disease state through “intragenic complementation” (the interaction of two mutations within the same gene that produces a novel functional effect) of other SCN5A mutations.13 In fact, several studies have indicated that some of these common polymorphisms may be informative clinically and relevant to the identification of those at risk for cardiac arrhythmias, particularly in the setting of TdP-inducing drugs or other environmental factors, as discussed later in this chapter.
Genotype-Phenotype Correlates in Long-QT Syndrome
The emergence of specific genotype/phenotype associations in LQTS suggest relatively gene-specific triggers, ECG patterns, and response to therapy (Fig. 32-3). Swimming- and exertion-induced cardiac events are strongly associated with mutations in KCNQ1 (LQT1), whereas auditory triggers and events occurring during the postpartum period most often occur in patients with LQT2. Whereas exertion- or emotional stress–induced events are most common in LQT1, events occurring during periods of sleep or rest are most common in LQT3. In a study population of 721 LQT1 and 634 LQT2 genetically confirmed patients from the U.S. portion of the international LQTS registry, a multivariate analysis was used to assess the independent contribution of clinical and mutation-specific factors to the occurrence of a first triggered event associated with exercise, arousal, or sleep/rest.14,15 Among the 221 symptomatic LQT1 patients, the first cardiac event was most often associated with exercise (55%), followed by sleep/rest (21%), arousal (14%), and nonspecific (10%) triggers; in contrast, the 204 symptomatic LQT2 patients most often had their first event associated with either arousal triggers (44%) or nonexercise/nonarousal triggers (43%), and only 13% of the symptomatic LQT2 patients had an exercise-triggered first event. In addition, males younger than 13 years with LQT1 had a nearly 3-fold increase in risk for exercise-triggered events, whereas females 13 years or older with LQT1 had a 3.5-fold increase in risk for sleep/rest nonarousal events. For LQT2 patients, the rate of arousal-triggered events was similar between boys and girls, but the rate of arousal-triggered events was significantly higher in women than in men (26% versus 6% at 40 years of age) after the onset of adolescence. Characteristic gene-suggestive ECG patterns have been described previously. LQT1 is associated with a broad-based T wave, LQT2 with a low-amplitude notched or biphasic T wave, and LQT3 with a long isoelectric segment followed by a narrow-based T wave.
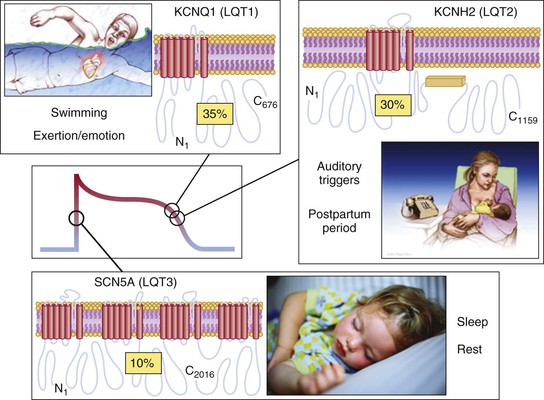
However, exceptions to these relatively gene-specific T wave patterns exist, and thus due caution must be exercised when making a pre–genetic test prediction of the particular LQTS subtype involved because the most common clinical mimicker of an LQT3-appearing ECG is seen in patients with LQT1. This is key to keep in mind because importantly, the underlying genetic basis heavily influences the response to standard LQTS pharmacotherapy (beta blockers), with beta blockers being extremely protective in LQT1 patients and moderately protective in patients with LQT2 and LQT3.16 Additionally, targeting the pathologic LQT3-associated late sodium current with agents such as mexiletine, flecainide, or ranolazine may represent a gene-specific therapeutic option for LQT3.17,18 Attenuation in repolarization with clinically apparent shortening of the QTc has been demonstrated with such a strategy, although no evidence-based survival benefit has been shown thus far.18 Realistically, however, at least a 30-year study may be needed for the latter. Even though the generalization that beta blocker efficacy depends on the genotype has been well accepted, the effectiveness of beta blocker therapy may be largely trigger-specific rather than dependent on genotype. In patients with either LQT1 or LQT2, beta blockade was associated with a pronounced 71% (LQT2 patients) to 78% (LQT1 patients) reduction in the risk for exercise-triggered cardiac events but had no statistically significant effect on the apparent risk for arousal- or sleep/rest-triggered events.4,15 However, it should be noted that many symptomatic LQT1 and LQT2 patients experience a subsequent cardiac event associated with a different trigger. For example, an LQT2 patient first experiencing an arousal event or one during sleep may subsequently have an exercise-triggered event. Therefore, beta blocker therapy still remains first-line therapy even for patients experiencing a non–exercise-associated first event.
In addition, intragenotype risk stratification has been completed for the two most common subtypes of LQTS based on mutation type, mutation location, and cellular function.19–22 Patients with LQT1 secondary to Kv7.1 missense mutations localizing to the transmembrane-spanning domains clinically have a twofold greater risk for an LQT1-triggered cardiac event than do LQT1 patients with mutations localizing to the C-terminal region. In addition, missense mutations localizing to the so-called cytoplasmic loops (C-loops) within the transmembrane-spanning domains, an area of the protein involved in the regulation of adrenergic channels, are associated with the highest rate of both exercise- and arousal-triggered events but not with an increase rate of sleep/rest-associated events.15 C-loop Kv7.1 missense mutations were consistently associated with greater than a sixfold increase in risk for exercise-triggered events in comparison to nonmissense mutations and an almost threefold increase in comparison to N- and C-terminal missense mutations.15
Patients with mutations resulting in a greater degree of Kv7.1 loss of function at the cellular in vitro level (i.e., dominant negative) have a twofold greater clinical risk than that of patients with mutations that damage the biology of the Kv7.1 channel less severely (haploinsufficiency). Adding to the traditional clinical risk factors, molecular location and cellular function are independent risk factors used in the evaluation of patients with LQTS.20 Akin to molecular risk stratification in LQT1, patients with LQT2 secondary to Kv11.1 pore region mutations have a longer QTc and more severe clinical manifestation of the disorder and experience significantly more arrhythmia-related cardiac events occurring at a younger age than do LQT2 patients with non–pore-related mutations in Kv11.1.23 Similarly, in a Japanese cohort of LQT2 patients, those with pore mutations had a longer QTc, and although not significant among probands, nonprobands with pore mutations experienced their first cardiac event at an earlier age than did those with a non–pore-related mutation.21 Most recently, additional information has been gleaned suggesting that LQT2 patients with mutations involving the transmembrane pore region had the greatest risk for cardiac events, those with frameshift/nonsense mutations in any region had intermediate risk, and those with missense mutations in the C-terminus had the lowest risk for cardiac events.22 Interestingly, LQT2 patients with mutations in the pore loop region of the Kv11.1 channel have a greater than twofold increased risk for arousal-triggered events, and LQT2 patients with non–pore loop transmembrane region mutations have an almost sevenfold increase in the risk for exercise-triggered cardiac events over patients with N-terminal/C-terminal (non-PAS domain) mutations.24
Incomplete penetrance and variable expressivity are the clinical hallmark features of LQTS, and it has long been thought that co-inheritance of a true disease-causing mutation and either a common or rare channel genetic variant may determine the expressed severity of the disorder. For example, coexistence of the common K897T-KCNH2 polymorphism and the A1116V-KCNH2 mutation (on opposite alleles) led to a more severe clinical course in a single Italian LQTS family. The A1116V mutation by itself produced a subclinical phenotype of mild QT prolongation and an asymptomatic course, whereas the proband hosting both variants had clinically overt disease consisting of diagnostic QT prolongation, presyncope episodes, and cardiac arrest.25 Besides cardiac ion channels, single nucleotide polymorphisms (SNPs) of non–ion channel genes such as NOS1AP (the gene encoding the nitric oxide synthase 1 adapter protein), ADRA2C (alpha2C-adrenergic receptor), and ADRB1 (beta1-adrenergic receptor) may modify disease severity in LQTS.26–29
In 2012, Amin and colleagues provided compelling evidence for a strong disease-modifying effect of a 3′ untranslated region (3′UTR), KCNQ1 allele–specific haplotype in LQT1 mutation–positive pedigrees; the magnitude of the effect on the QTc and symptomatology goes well beyond any other currently described genetic modifiers.30 The KCNQ1 gene encodes for a single Kv7.1 ion channel alpha subunit. Following KCNQ1 gene expression and post-translational modifications, four alpha subunits are assembled to create a pore-forming Kv7.1 tetrameric channel. Therefore, if a patient had a heterozygous KCNQ1 mutation (i.e., one normal KCNQ1 gene allele and one mutant allele), one would expect that if both the normal and mutant gene alleles were expressed in equal amounts, 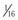 of the channels would be normal homomeric tetramers and
of the channels would be normal homomeric tetramers and 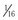 would be mutant homomeric tetramers. The remaining channels would be hybrids containing both normal and mutant alpha subunits. One would predict that if the normal KCNQ1 gene allele expression was somehow suppressed, there would be relatively more KCNQ1 mutant alpha subunits translated and ultimately assembled to provide more dysfunctional KCNQ1 channels and thus lead to a more severe manifestation of the disorder (Fig. 32-4). Simply put, far more bad (mutant) channels than good (healthy) channels would be created. The opposite would be true if the mutation containing the KCNQ1 allele were suppressed.
would be mutant homomeric tetramers. The remaining channels would be hybrids containing both normal and mutant alpha subunits. One would predict that if the normal KCNQ1 gene allele expression was somehow suppressed, there would be relatively more KCNQ1 mutant alpha subunits translated and ultimately assembled to provide more dysfunctional KCNQ1 channels and thus lead to a more severe manifestation of the disorder (Fig. 32-4). Simply put, far more bad (mutant) channels than good (healthy) channels would be created. The opposite would be true if the mutation containing the KCNQ1 allele were suppressed.
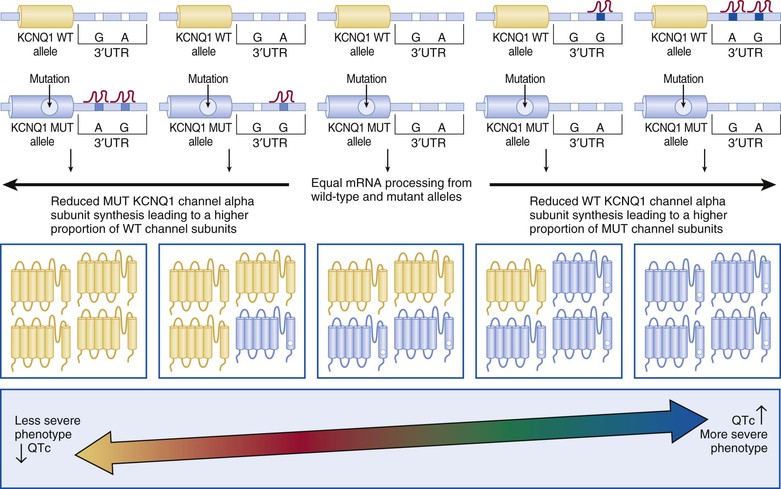
Most genes have a 3′UTR that produces an mRNA transcript that contains regions of cis-regulatory binding sites for small noncoding microRNA (miRNA) molecules that bind to the transcript and ultimately inhibit that gene’s expression. Naturally occurring genetic variation within these 3′UTRs (miR-SNPs) can either abolish existing or create new miRNA binding sites. Amin and colleagues identified three naturally occurring SNPs (rs2519184, rs8234, and rs10798) within the KCNQ1 3′UTR whereby the presence of their minor alleles (A, G, G) generates a “suppressive” haplotype by creating new miRNA binding sites that suppress expression of the KCNQ1 gene allele in which they reside.30 In a cohort of 168 KCNQ1 (LQT1) mutation–positive individuals from 41 families, Amin and colleagues showed that inheritance of the “suppressive” haplotype residing on the normal “healthy” allele produced a more severe LQT1 phenotype with regard to QTc and symptomatology than did inheritance of the “suppressive” haplotype residing on the same allele as the KCNQ1 mutation (shorter QTc and fewer symptoms).30 This intriguing discovery may not only explain a significant component of the reduced penetrance and variable expressivity that is a common feature of arrhythmia syndromes but may also represent a paradigm shift in our thinking about disease-modifying genetic drivers of mendelian disorders because one of the most important genetic determinants of disease severity in LQT1 appears to be the 3′UTR KCNQ1 haplotype on the allele inherited from the unaffected “non-LQTS” parent.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


