Genetic Studies of Myocardial and Vascular Disease
Qing Wang
Reed E. Pyeritz
Christine E. Seidman
Craig T. Basson
Overview
Genetic factors contribute to the pathogenesis of a wide variety of disorders that affect cardiac and vascular structure and function. Advances in molecular genetics since the 1980s have provided remarkable insights into the genes responsible for and the pathogenic mechanisms of cardiovascular disease. Disease-causing genes have been identified for cardiac arrhythmic disorders (e.g., long-QT syndrome [LQT], Brugada syndrome, and idiopathic ventricular fibrillation [IVF]), congenital heart disease (e.g., Holt-Oram syndrome, Ellis-van Creveld syndrome, atrial septal defect [ASD] with atrioventricular [AV] block, cardiac myxoma, hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic right ventricular dysplasia, and familial vascular anomalies (e.g., Marfan syndrome [MFS], patent ductus arteriosus, supravalvular aortic stenosis, lymphedema). Genetic studies of human cardiovascular disorders continue to provide insight into molecular mechanisms underlying morphogenesis, development, and physiologic function of the cardiovascular system. Molecular genetic analysis of cardiovascular disease permits DNA-based diagnosis, which will promote new approaches to risk stratification and therapy. These exciting molecular discoveries have raised the hope of genetic testing, drug development, and gene-based therapies for a variety of cardiovascular disorders. In this chapter, we discuss major advances in the genetics of myocardial and vascular disease.
Glossary
Allele
Alternative forms of a gene or a genetic locus.
Allelic
A condition in which two phenotypically different traits are the result of different mutations in the same gene.
Compound heterozygote
An individual with one mutation in one allele of a given gene and a different mutation in the other allele of the gene. Compound heterozygous mutations are usually associated with autosomal recessive disease.
Heterogeneity
Allelic or intragenic heterogeneity refers to the existence of different genotypes at the same genetic locus that cause a similar phenotype. Locus heterogeneity refers to a situation in which a disease is caused by defects at different genetic loci.
Heterozygote
An individual with a mutation in one copy of a given gene but without a mutation in the other copy. Heterozygous mutations are usually associated with autosomal dominant disease.
Homozygote
An individual with the identical sequence in both alleles of a given gene. Homozygous mutations are usually associated with autosomal recessive disease.
Linkage analysis
Analysis to determine whether two or more genetic traits (a marker locus and a disease trait) are cosegregating within a pedigree.
Positional cloning
A technique used to identify the underlying genetic cause of a disease based primarily on finding the location of the defective gene.
Introduction
Cardiovascular disease is the leading cause of death in developed countries. Genetic factors play an important role in the pathogenesis of cardiovascular disease. Each human being is variably estimated to have approximately 25,000 different genes (1). Mutations in single or multiple genes can cause cardiovascular disease. The revolutionary advances in molecular genetics over the last 20 years have provided new insights into the responsible genes and pathogenic mechanisms of cardiovascular disease. Some of these diseases are individually quite prevalent. Many of the most common diagnoses, such as “stroke,” “heart attack,” “hypertension,” or “heart failure,” subsume several distinct disorders. Other cardiovascular conditions are quite rare but have been instructive in terms of the etiology and pathogenesis of common disease processes, such as atherosclerosis, aneurysms, and dysrhythmia. Mendelian disorders may be rare and can be caused by mutations in a single gene with profound consequences. Many of the more common disorders are not simply inherited and have a few or perhaps many genes that stochastically interact with each other and with the environment to produce pathology.
Monogenic cardiovascular disorders can be inherited as autosomal dominant, autosomal recessive, or X-linked traits. A rapidly increasing number of the genes responsible for such disorders have been mapped to specific chromosomal regions by linkage analysis and identified by candidate gene or positional cloning approaches (2,3).
Analysis of the genetics of common disease is far more complicated, whether in terms of populations, families, or molecules, than that for simply inherited conditions. Nonetheless, considerable progress has been made, both in experimental animals and in humans, in identifying genes that contribute to the cause, pathogenesis, or both, of common diseases such as coronary artery disease, myocardial infarction, and hypertension. In many instances, specific genetic variations that confer an increased risk of developing a condition can now be identified before symptoms develop. The challenge is to devise approaches to modulating the risk in those so identified.
This chapter reviews the major advances in the molecular genetics of single-gene disorders of the cardiovascular system. The genetics of complex diseases, such as coronary artery disease and hypertension, is covered in other chapters.
Disorders of Cardiac Rhythm and Conduction
Cardiac arrhythmias are a common cause of morbidity and mortality, accounting for 300,000 to 400,000 sudden deaths each year in the United States alone (4,5). A number of Mendelian conditions account for an unknown but clearly underestimated fraction of this mortality.
Long-QT Syndrome
LQT is characterized by prolongation of the QT interval and T-wave abnormalities on electrocardiograms (ECGs) and symptoms of syncope, seizures, and sudden death, usually from a specific ventricular arrhythmia, torsades de pointes (6,7,8). Two inherited forms of LQTS have been reported: autosomal dominant LQTS (Romano-Ward syndrome) with normal hearing (6,8) and the much rarer autosomal recessive LQTS (Jervell-Lange-Nielsen syndrome) associated with congenital deafness (6,9). It appears that patients with Jervell-Lange-Nielsen syndrome have a more adverse course and higher rate of sudden death. The primary defect at the tissue level involves cardiac repolarization, and the prolonged QT interval, when corrected for heart rate, is usually (but not always) evident on standard 12-lead ECG (Fig. 96.1). Autosomal dominant LQTS has an incidence of approximately 1 per 10,000, without apparent ethnic or geographic predilection (10,11). A set of clinical criteria to assist in diagnosis has been developed (Fig. 96.1) (12). Although most clinical events are “triggered” by excitement, exercise, emotional upset, and the like, sudden death can occur during sleep. Some have suggested LQTS as a cause of sudden infant death syndrome (13,14).
Molecular Genetics
Molecular genetic studies have identified nine genes for LQTS alone or syndromes involving LQTS and other diseases (discussed in detail later). These genes include KCNQ1 or KVLQT1 on chromosome 11p15.5 (LQT1), KCNH2 or HERG on 7q35–36 (LQT2), SCN5A on 3p21–24 (LQT3), ANK2 on 4q25–27 (LQT4), KCNE1 on 21q22 (LQT5), KCNE2 on 21q22 (LQT6), KCNJ2 on 17q23–24 (LQT7, Andersen syndrome), HCN4 on 15q24–q25 (LQT8, sick sinus syndrome), and CACNA1C on 12p13.3 (LQT9, Timothy syndrome). Heterozygous mutations in these genes cause autosomal dominant LQTS. Homozygous or compound heterozygous mutations in KVLQT1 and KCNE1 cause Jervell-Lange-Nielsen syndrome (LQTS associated with deafness). Mutations in KCNQ1, KCNH2, and SCN5A account for more than 95% of genotyped patients with LQTS, and mutations in other genes are rare.
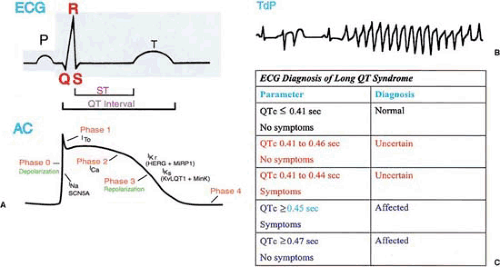 FIGURE 96.1. A: Temporal relationship between the electrocardiogram (ECG) and the cardiac action potential (AC). B: A representative ECG pattern for torsades de pointes (TdP). C: Conservative diagnostic criteria for long-QT (LQTS) syndrome. This diagnostic system has been successful in identification of LQTS genes (18). |
Cardiac Potassium Channel Gene KCNQ1
KCNQ1, the LQT1 gene on chromosome 11p15.5, is the first novel gene cloned for LQTS (15). This gene encodes a potassium channel α-subunit with a conserved potassium-selective pore-signature sequence flanked by six transmembrane-spanning segments (S1 to S6). People who are heterozygous for mutations in KCNQ1 are at risk for autosomal dominant LQTS. However, people who have mutations in both of their KCNQ1 alleles have the more severe disease, autosomal recessive LQTS with congenital deafness (16,17). Mutations in KCNQ1, when heterozygous, cause loss of channel function, altered channel gating, and/or a dominant-negative effect (18) in which the mutant form of the channel subunit interferes with the function of the normal form. The KCNQ1 protein normally functions by interacting with MinK (encoded by Kenel) a short potassium channel subunit with a mere 130 amino acids and only one transmembrane-spanning segment (19,20). The physical interaction between KCNQ1 and MinK produces the slowly activating potassium current (IKs) in cardiac myocytes.
Various ion currents such as IKs are responsible for the different phases of the cardiac action potential (CAP) (Fig. 96.1). One CAP corresponds to one heartbeat. The time course of
one CAP can be divided into five phases: upstroke of rapid depolarization (phase 0); rapid repolarization following the peak (phase 1); a plateau (phase 2); rapid repolarization (phase 3); and the period between the maximum negativity (maximum diastolic potential) and the upstroke of the next action potential (phase 4) (Fig. 96.1) (21,22). The IKs potassium current acts at the repolarization phase (phase 3). KCNQ1 mutations reduce the repolarizing cardiac IKs current, causing delayed repolarization and prolongation of the CAP duration and prolongation of the QT interval on ECG.
one CAP can be divided into five phases: upstroke of rapid depolarization (phase 0); rapid repolarization following the peak (phase 1); a plateau (phase 2); rapid repolarization (phase 3); and the period between the maximum negativity (maximum diastolic potential) and the upstroke of the next action potential (phase 4) (Fig. 96.1) (21,22). The IKs potassium current acts at the repolarization phase (phase 3). KCNQ1 mutations reduce the repolarizing cardiac IKs current, causing delayed repolarization and prolongation of the CAP duration and prolongation of the QT interval on ECG.
Cardiac Potassium Channel Gene KCNH2
The LQT2 gene on chromosome 7q (23,24) was identified as KCNH2, which encodes a cardiac potassium channel core–forming subunit with six transmembrane segments (25). This channel ordinarily generates the rapidly activating delayed rectifier potassium current (IKr) in the heart. LQTS-associated mutations in KCNH2 act through either a loss-of-function or a dominant-negative mechanism (26). The IKr potassium current is a major current at the repolarization phase of CAP. KCNH2 mutations reduce the repolarizing cardiac IKr current, thus causing prolongation of the CAP duration, leading to prolongation of the QT interval on ECG.
Cardiac Sodium Channel Gene SCN5A
The gene associated with LQT3 on chromosome 3p is the cardiac sodium channel gene, SCN5A (11,27). It encodes a large protein of 2,016 amino acids with a putative structure of four homologous domains (DI to DIV), each of which contains six membrane-spanning segments (S1 to S6) (28). The sodium current INa generated by this channel is responsible for the depolarization phase of the CAP and contributes some current at the plateau phase. LQTS-causing mutations in SCN5A act through a gain-of-function mechanism in which the mutant channel functions but with altered properties. Some SCN5A mutations (Δ KPQ, N1325S, R1644H, E1784K) generate the persistent noninactivated sodium current in the plateau phase of the CAP (23,24). One SCN5A mutation (D1790G) shifts steady-state inactivation by –16 mV in the presence of the β1-subunit (29). The E1784K mutation shifts the voltage dependence of steady-state inactivation toward more negative potentials (5 mV), and coexpression with the β1-subunit exaggerates the negative shift. The mutation R1623Q increases the probability of long openings and causes early reopenings, producing a threefold prolongation of sodium current decay (30,31). Although the LQTS-causing mutations in SCN5A produce variable degrees of altered channel function, almost all of them lead to the increase of inward plateau sodium current, prolonging the action potential duration and the QT interval on ECG.
Membrane Protein Adaptor Gene Ankyrin-B (ANK2 or ANKB)
The LQT4 gene was mapped onto chromosome 4q25–27 in a large French family with autosomal dominant LQTS associated with sinus node dysfunction (bradycardia) and atrial fibrillation (32). The disease-causing mutation in the French kindred was identified as a loss-of-function mutation in ankyrin-B (33), establishing ANK2 as the LQT4 gene. Several other ANK2 loss-of-function mutations were identified in patients with varying cardiac dysfunction including bradycardia, sinus arrhythmias, idiopathic ventricular fibrillation, catecholaminergic polymorphic ventricular tachycardia, and risk of sudden death (34). Some of ANK2 mutation carriers do not have prolonged QTc on ECG (34). Ankyrins are a family of adaptor proteins that link integral membrane proteins to spectrin-based cytoskeleton (35). Ankyrins bind to several ion channel proteins, such as anion exchanger, Na/K-ATPase, voltage-sensitive sodium channel (INa), and Na/Ca exchanger (INa-Ca), and calcium-release channels including inositol 1,4,5-trisphosphate (IP3) and ryanodine receptors (35). Expression of α1, α2 Na/K-ATPase, Ncx (INa-Ca), and IP3 receptors was reduced in adult AnkB+/- murine myocytes (33). The combined reduction in the expression of these ankyrin-B–interacting
proteins may trigger the abnormal electrophysiologic and Ca2+ dynamics in AnkB+/- cardiomyocytes, leading to arrhythmias (35).
proteins may trigger the abnormal electrophysiologic and Ca2+ dynamics in AnkB+/- cardiomyocytes, leading to arrhythmias (35).
Cardiac Potassium Channel Gene KCNE1
As in KCNQ1, heterozygous mutations in KCNE1 cause autosomal dominant LQTS, but homozygous or compound heterozygous mutations cause autosomal recessive LQTS (36). The mink protein encoded by Kenel interacts with KCNQ1 and KCNH2, recapitulating IKs with KCNQ1 and augmenting the amplitude of IKr generated by KCNH2 (37). Different MinK mutations appear to have different effects on KCNQ1 and KCNH2 (37). Mutation V47F alters IKs currents and increases the KCNH2 current. W87R alters IKs currents only. D76N suppresses both KCNQ1 and KCNH2 currents. Mutant L51H channels are processed improperly and interact with neither KCNQ1 nor KCNH2.
Cardiac Potassium Channel Gene KCNE2
This gene encodes MiRP1, for MinK-related peptide 1, which is an integral component of a potassium channel. This gene has high homology with KCNE1; their close linkage (70 kb apart on chromosome 21q22) suggests that one arose as a result of gene duplication of the other. Mutations in KCNE2 cause LQT6 (38). MiRP1 is a small integral membrane subunit that assembles with KCNH2 to form IKr. MiRP1 was also shown to interact with KCNQ1, resulting in a great change of the amplitude and gating properties of the KCNQ1 current (39).
Genotype–Phenotype Correlation in Long-QT Syndrome
Mutations in KCNQ1 and KCNH2 cause approximately 40% of LQTS cases, respectively, whereas mutations in SCN5A are associated with 10% of cases (40). Patients with LQT1 tend to have frequent exercise-related cardiac events, whereas most patients with LQT3 experience cardiac events during sleep or at rest. Patients with LQT2 may experience cardiac events both at rest and during exercise. The risk of cardiac events is higher in LQT1 and LQT2 than in LQT3; however, the likelihood of dying during a cardiac event is significantly higher among LQT3 patients (41). LQT1, LQT2, and possibly LQT3 genotypes are associated with different but typical ST-T–wave patterns on ECG (42).
Diagnosis, Genetic Testing, and Treatment
LQTS can be diagnosed with reasonable certainty if QTc is 0.47 second or longer for asymptomatic individuals or if QTc is 0.45 second or longer for symptomatic individuals (21,22). LQTS is not likely if QTc is less than 0.41 second in male and less than 0.43 second in female patients. Many mutation carriers as well as normal people have QTc ranging from 0.41 to 0.46 second, which significantly limits the power of ECG diagnosis of LQTS patients. Sudden death occurs in 10% to 30% symptomatic LQTS patients (10), and it may occur with the first syncope. These factors make genetic testing extremely important. Genetic testing for LQTS patients is available commercially. Only positive results—that is, identification of a mutation—confirm the diagnosis.
The identification of LQTS genes has led to the development of gene-specific therapies for LQTS patients (22). Sodium channel–blocking agents such as mexiletine and flecainide can shorten QTc for LQT3 patients (43,44). Raising the serum potassium concentration shortens QTc for LQT2 patients (45). Potassium channel–opening agents and verapamil may be beneficial to patients with potassium channel mutations (10). All these therapies are at the investigational stage, and it is not clear whether these therapies can eliminate life-threatening ventricular arrhythmias.
All symptomatic patients as well as all asymptomatic children require treatment (10). For most LQTS patients, administration of a β-blocker is the initial choice of therapy. β-Blockers are effective in patients with KVLQT1 and HERG mutations. Other therapeutic options include pacing and implantation of a cardioverter-defibrillator in combination with β-blockers.
Andersen Syndrome
Mutations in the Kir2.1 inward rectifier potassium channel were identified in patients with Andersen syndrome, which is characterized by QTc prolongation, periodic skeletal muscle paralysis, and mild dysmorphic features (46). Kir2.1 mutations act by either a loss-of-function mechanism or a dominant-negative mechanism. Kir2.1 consists of only two transmembrane segments, M1 and M2, flanking a pore region and can generate the IK1 potassium current at the terminal repolarization phase (phase 3) and for maintenance of the cardiac resting potential.
Sick Sinus Syndrome
Sick sinus syndrome (SSS) represents a variety of arrhythmic conditions caused by sinus node dysfunction. Patients with SSS display sinus bradycardia, sinus arrest, and/or sinoatrial block on ECG and manifest syncope, presyncope, dizziness, and fatigue. Episodes of atrial tachycardias coexisting with sinus bradycardia (“tachycardia-bradycardia syndrome”) can also be found in this disorder. Compound heterozygous mutations in the cardiac sodium channel gene SCN5A cause autosomal recessive SSS (47). All these SCN5A mutations lead to loss of function or significant impairments in channel gating (inactivation).
In a patient with idiopathic SSS, a heterozygous 1-bp deletion (1631delC or HCN4–573X) in exon 5 of the human HCN4 (hyperpolarization-activated cyclic nucleotide-gated channel 4) gene on chromosome 15q24–q25 was identified (48). The HCN4 gene encodes a potassium channel with six putative transmembrane segments and is responsible for the cardiac pacemaker current I(f), a major determinant of diastolic depolarization in sinus nodal cells. Mutant HCN4-573X channels are insensitive to increased cellular cAMP levels and have a dominant-negative effect. In a separate study, an HCN4 missense mutation, D553N, was identified in a patient with sinus node dysfunction who showed recurrent syncope, LQTS, and polymorphic ventricular tachycardia, torsades de pointes (49). Mutant HCN4 channel with the D553N mutation reduces trafficking to the cell membrane and acts by a dominant-negative mechanism (49).
Timothy Syndrome
Some LQTS patients also display webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and autism, and this type of LQTS has been named Timothy syndrome. Two mutations, G402S and G406R in exon 8A, were identified in the CACNA1C gene encoding cardiac L-type calcium channel (CaV1.2) (50). Both mutations reduced channel inactivation (50).
Short-QT Syndrome
Gaita et al. described a new syndrome called short-QT syndrome (SQTS) (51). Patients with SQTS have QTc of 300 msec
or less on baseline ECG and exhibit syncope, palpitations, resuscitated cardiac arrest, and a positive family history for sudden cardiac death. Mutations in three genes have been identified in patients with SQTS. One KCNH2 mutation, N588K, was identified in two families with SQTS (52). The KCNH2 mutation is a gain-of-function mutation that dramatically increases the IKr potassium current. The second gene for SQTS has been identified as KCNQ1. A KCNQ1 mutation, the V307L mutant, was identified in a patient with SQTS and is a gain-of-function mutation (53). The mutation D172N in KCNJ2 was identified in a small family with two patients, and it increases the outward IK1 potassium current (54).
or less on baseline ECG and exhibit syncope, palpitations, resuscitated cardiac arrest, and a positive family history for sudden cardiac death. Mutations in three genes have been identified in patients with SQTS. One KCNH2 mutation, N588K, was identified in two families with SQTS (52). The KCNH2 mutation is a gain-of-function mutation that dramatically increases the IKr potassium current. The second gene for SQTS has been identified as KCNQ1. A KCNQ1 mutation, the V307L mutant, was identified in a patient with SQTS and is a gain-of-function mutation (53). The mutation D172N in KCNJ2 was identified in a small family with two patients, and it increases the outward IK1 potassium current (54).
Brugada Syndrome
Idiopathic ventricular fibrillation (IVF) is ventricular fibrillation without underlying structural heart disease and other electrolyte disorders such as LQTS. IVF can be classified into two types: IVF with normal ECG and IVF with an ECG feature of right-bundle-branch block and persistent ST-segment elevation (STE) in leads V1 to V3 (IVF with STE) (Fig. 96.2), which is now often referred to as Brugada syndrome (55,56,57). In a small percentage of patients with IVF, a family history of sudden death or even IVF can be found, with inheritance as an autosomal dominant pattern (58).
In 1998, the first gene for IVF with STE (Brugada syndrome) was identified as SCN5A




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
