Genetic Factors in Atherosclerosis
Aldons J. Lusis
Overview
Mendelian disorders with large effects are the most dramatic examples of genetic contributions to atherosclerosis, the primary cause of coronary artery disease (CAD) and myocardial infarction (MI). Common forms of the disease, however, are the product of many genes, each with modest effects, that are modified by the environment, rather than a single, highly penetrant gene.
Genetic factors can influence atherosclerosis and its clinical sequelae in many ways and at different stages. A significant fraction of genetic susceptibility can be explained by effects on risk factors such as elevated cholesterol, elevated blood pressure, and reduced high-density-lipoprotein levels. Genetic differences also appear to influence inflammatory cells, vessel wall functions, plaque stability, and thrombosis.
Genetics is important for two reasons. First, it can lead to an understanding of the molecular bases of the disease, providing novel therapeutic targets. Second, genetic studies have the potential to reveal the nature of the individual differences that contribute to genetic susceptibility. Studies of Mendelian disorders and of transgenic animal models have revolutionized our understanding of pathways involved in atherosclerosis. Little is known of genetic interactions involved in common forms of the disease, but as our understanding increases, it may be possible to develop more-individualized, genetics-based, therapies.
Glossary
Allele
One alternative form of a gene.
Candidate gene
A gene that, by virtue of its known properties, could contribute to a genetic disease.
Genetic marker
A locus that has readily classifiable alleles that can be used in genetic studies, such as a restriction fragment length polymorphism or a single-nucleotide polymorphism.
Genotype
Either the genetic constitution of an individual or the alleles that are present at a particular locus.
Haplotype
A particular combination of allelic DNA variations at a locus on a chromosome.
Heritability
The proportion of variance of a trait (such as blood pressure or myocardial infarction) explained by genetics.
Linkage
The tendency of genes that reside near one another on a chromosome to be inherited together.
Locus
A unique chromosomal location that defines the position of a gene.
Lod score
A statistical method that tests genetic marker data in families to determine whether two loci are linked.
Missense mutation
A mutation that changes a codon for one amino acid to another amino acid.
Modifier gene
A gene whose expression can modulate a phenotype.
Multifactorial
A trait determined by multiple factors, usually including genetic and environmental factors.
Nonsense mutation
A mutation that results in the introduction of a chain-termination codon.
Penetrance
The probability that an individual carrying a disease allele will exhibit the disease.
Phenotype
The physical characteristics of an individual determined by his or her genetic constitution (e.g., the phenotype of high levels of plasma cholesterol results from mutations in low-density-lipoprotein receptor).
Polymorphism
A genetic variation that occurs relatively frequently (>1%) in the population.
Single-nucleotide polymorphism (SNP)
The most common variety of genetic variation in the population, involving the substitution of one base for another.
Mendelian Traits Associated with Coronary Artery Disease
More than 60 Mendelian mutations have been associated with CAD and many more with hypertension and diabetes (1). A partial list of such disorders is presented in Table 95.1. Most of these disorders are quite rare, with a population frequency of less than one in tens of thousands, although some, such as familial hypercholesterolemia (FH) and familial defective apoB-100, occur with a frequency of more than 1 in 1,000 in many populations.
The classic genetic trait in heart disease is FH, first described by Carl Muller in Norway in the 1930s (2). He noted that the triad of highly elevated cholesterol, tendon xanthomas, and early heart disease segregated together as a dominant trait, paving the way for subsequent large epidemiologic studies that showed a clear relationship between elevated cholesterol and CAD. About 30 years later, Joseph Goldstein and Michael Brown initiated detailed molecular studies of the disease, demonstrating that it results from various defects in the realization of the low-density-lipoprotein (LDL) receptor (3). FH heterozygotes, with one mutant copy of the LDL receptor gene, typically have plasma cholesterol levels ranging from 300 to 550 mg/dL, whereas FH homozygotes, with two mutant copies, typically have cholesterol levels ranging from 550 to greater than 1,000 mg/dL (Fig. 95.1). FH homozygotes usually develop severe CAD by their teens, but the penetrance of CAD in heterozygotes varies widely and depends on the same lifestyle modifiers as in noncarriers, including diet, smoking, and activity level (4). FH provides a beautiful exemplar of the importance of studies of rare disorders for understanding the more common forms. Studies of FH provided strong evidence for a direct role of elevated LDL in CAD. The molecular studies of FH also led to an understanding of the nature of cholesterol homeostasis, and this, in turn, was a major impetus for the development of cholesterol-lowering drugs (3).
Several different gene defects have now been recognized that depress LDL receptor function and thereby elevate cholesterol levels. A rare autosomal recessive form of hypercholesterolemia (ARH) results from mutations of an LDL receptor adaptor protein. Patients with ARH have normal expression of LDL receptor but greatly reduced clearance of circulating LDL by liver. The adapter protein, named ARH, is required for efficient endocytosis of the LDL receptor in selected tissues (5). A novel dominant form of hypercholesterolemia, very similar in cholesterol levels to FH, results from gain-of-function, missense mutations of the PCSK9 gene, which encodes neural apoptosis-regulated convertase (NARC-1) (6). Experimental studies indicate that the protein results in a downregulation of the LDL receptor. Of broader significance, common nonsense mutations of the PCSK9 gene have proved to be a common cause of reduced cholesterol levels (7).
Familial defective apoB100 (FDB) is due primarily to a relatively common mutation in apolipoprotein B that substitutes glutamine for arginine at amino acid 3500, impairing the ability of LDL to bind to the LDL receptor. Heterozygous individuals exhibit cholesterol levels of about 300 mg/dL owing to defective clearance, and, unlike FH, the patients do not have tendonous xanthomas (8,9).
Sitosterolemia is a rare disorder associated with greatly increased absorption of plant sterols and with hypercholesterolemia. The disease results from mutations of either of two ABC half-transporters, ABCG5 and ABCG8, that are responsible for transport of sterols, particularly plant sterols, out of enterocytes into the intestinal lumen. The transporters also play a role in the transport of sterols into the bile (9,10).
Mutations of lipoprotein lipase or its cofactor apoCII result in recessive familial forms of hypertriglyceridemia. Although these are characterized by massive accumulation of plasma triglyceride-rich lipoproteins, the effects on atherosclerosis appear to be modest (11).
High-density-lipoprotein (HDL) levels are inversely correlated with atherosclerosis. The primary Mendelian disorders associated with reduced HDL levels include deficiencies of apolipoprotein AI, the major structural protein of HDL, and ABCA1, a phospholipid/cholesterol transporter largely responsible for cholesterol efflux from intestine or liver to apoAI. Heterozygous carriers of these disorders have significantly reduced HDL levels, and homozygous individuals are characterized by the near absence of HDL-cholesterol levels and early CAD (12,13,14). Deficiencies of lecithin–cholesterol acyl transferase (LCAT) also result in decreased HDL levels, whereas loss-of-function mutations of cholesterol ester transfer protein (CETP) result in elevated HDL levels (Fig. 95.1) (11).
The mutations for a number of Mendelian forms of hypertension have been identified (Table 95.1). For the most part, these mutations impair the handling of renal salts, providing a molecular understanding of the pathogenesis of hypertension (15). I do not discuss the genetics of hypertension in depth in this review because it is the subject of a separate chapter in this book.
A structural variant of the coagulation factor V (Arg500Gln) known as Factor V Leiden is a major cause of thromboembolic disease in European populations, where it has an allele frequency of several percent. The disorder exhibits incomplete penetrance, and about 80% of homozygous patients and 10% of heterozygous patients will have thrombosis in their lifetimes. Factor V Leiden increases the risk of myocardial infarction, stroke, and thrombosis in men (16,17).
Homocystinuria, a rare autosomal recessive disorder, is characterized by highly elevated levels of homocysteine in the plasma and a high frequency of occlusive vascular disease. It is due primarily to mutations in the cystathionine β-synthetase (CBS) gene, although deficiencies of methylenetetrahydrofolate reductase (MTHFR) or enzymes in cobalamin metabolism can also elevate homocysteine. Based on the findings with homocystinuria, slightly elevated homocysteine levels were found to be significantly associated with atherosclerosis in the general population (18,19).
As discussed later, insulin resistance and type 2 diabetes (T2D) are strongly associated with CAD. Most forms of T2D are multifactorial, but several rare Mendelian forms have been characterized (Table 95.1). Some of the genes involved in Mendelian forms also appear to contribute to polygenic T2D. For example, rare missense or nonsense mutations of the PPARg, KCNJ11, HNF4A, HNF1A, and INS genes result in severe diabetes or lipodystrophy, and common polymorphisms of these genes have also been associated with common T2D (20).
Wang and colleagues (21) recently mapped a locus contributing a dominant form of CAD and MI in a large pedigree and identified the transcription factor MEF2A as a likely
candidate based on the fact that the affected individuals shared an uncommon 21-base pair deletion of a gene. Subsequently, population screening of control subjects and subjects with CAD revealed mutations of the gene in both unaffected and affected individuals, raising the possibility that a gene other than MEF2A at the locus was responsible for the disease. Additional population studies and experimental studies will be required to resolve the issue.
candidate based on the fact that the affected individuals shared an uncommon 21-base pair deletion of a gene. Subsequently, population screening of control subjects and subjects with CAD revealed mutations of the gene in both unaffected and affected individuals, raising the possibility that a gene other than MEF2A at the locus was responsible for the disease. Additional population studies and experimental studies will be required to resolve the issue.
TABLE 95.1 Mendelian Disorders Relevant to Atherosclerosis | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
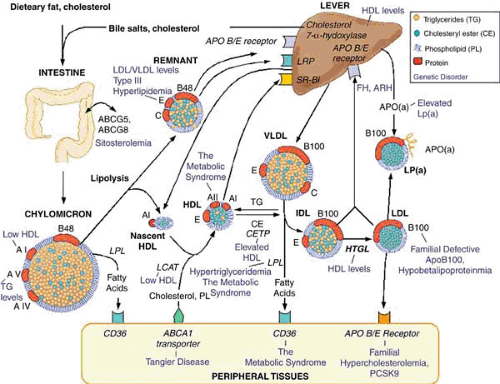 FIGURE 95.1. Candidate genes in lipoprotein metabolism. Lipids are transported through the circulation as complexes with various apolipoproteins that serve to package lipids and act as cofactors for enzymes or ligands for uptake by cellular receptors. Dietary lipids are absorbed in the intestine, packaged into chylomicrons, and secreted into the lymph. On entering the circulation, the triglycerides are hydrolyzed through the action of lipoprotein lipase (LPL), and the resulting remnants are taken up by the interaction of apolipoprotein E (apoE) with the LDL receptor (apoB, E receptor) or the LDL-receptor–related protein (LRP). During lipolysis, the surface phospholipids and proteins of chylomicrons slough off to give rise to HDL precursors. Liver cells package triglycerides and cholesterol esters into very-low-density-lipoprotein (VLDL) particles. These are acted on by LPL to produce intermediate-density lipoproteins (IDLs), which can be taken up by the apoB, E receptor or further lipolyzed, partly through the action of hepatic lipase (HL) to produce LDL. LDLs retain a single major protein, apoB100, and are removed from the circulation by the apoB, E receptor. Because of the slow kinetics of LDL uptake, LDL particles are the major cholesterol-carrying particles in most individuals. LDL can complex with a protein termed apo(a) [APO(a)] to form lipoprotein (a) [LP(a)] particles, which appear to be particularly atherogenic. HDLs are formed largely in the circulation from apolipoproteins AI and AII secreted by liver and intestine and from the surface of chylomicrons and VLDL during their lipolysis. The HDL precursors take up cholesterol from various tissues through interaction with the ABCA1 transporter, and the cholesterol is esterified by the action of lecithin–cholesterol acyl transferase (LCAT). Lipids can be transferred between lipoproteins through the action of cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP). ARH, autosomal recessive hypercholesterolemia; FH, familial hypercholesterolemia; HTGL, hepatic triglyceride lipase. (Modified Lusis AJ, Fogelman AM, Fonarow GC. Genetic basis of atherosclerosis. Part I: new genes and pathways. Circulation 2004;110:1868–1873. ) |
A striking example of a disorder that results in early-onset CAD independent of other known risk factors is Hutchinson-Gilford progeria. This rare Mendelian disease is characterized by extremely accelerated aging, and death usually results from myocardial infarction (MI) at a median age of 13 year. Recent studies have shown that the disease is due to a specific mutation at the lamin A gene, whose product is associated with the nuclear envelope. A variety of mutations of this gene result in very different phenotypes, including familial partial lipodystrophy (22,23).
As discussed later, studies in experimental animals and humans suggest that genes affecting blood cells and vascular cells are likely to be important determinants of CAD susceptibility. Thus, there are likely to be other highly penetrant genes
influencing atherosclerosis that do not affect the common risk factors. Given the late age of onset of the clinical disease, their identification will be a major challenge.
influencing atherosclerosis that do not affect the common risk factors. Given the late age of onset of the clinical disease, their identification will be a major challenge.
Common Forms of Atherosclerosis Have a Strong Genetic Component
With the exception of Mendelian disorders, CAD and cerebrovascular disease do not exhibit obvious genetic patterns of inheritance. Because the diseases are so common, it is not surprising that there are frequently multiple affected individuals in families. What is the evidence that the common forms of CAD, MI, and stroke exhibit a significant genetic component?
Statistical models intended to explain the inheritance of common traits were first developed in 1918 by Fisher (24). Whereas traits governed by a small number of loci (i.e., Mendelian traits) often yield discontinuous distributions (such as cholesterol levels in FH individuals as compared to the rest of the population), Fisher postulated that most common traits result from the additive interactions of a large number of loci as well as environmental influences. In the case of quantitative traits, such as blood pressure, cholesterol levels, and body mass index, this would yield the observed normal, or Gaussian, distributions in the population. Such distributions are specified by two parameters—the mean and the variance. The variance is a statistic used to quantify the dispersion about the mean. When variances are due to independent causes, they are additive. Thus, the overall variance of a phenotype (VP), such as cholesterol levels or blood pressure, is the sum of the individual causes. These can be divided into the environmental variance (VE) and the genetic variance (VG). The heritability (h2) of a trait is the proportion of total variance that is genetic (VG/VP). Heritabilities of human traits can be estimated from family studies or twin studies (see later discussion). The genetic variance VG can be partitioned into components with additive, dominant, and epistatic influences (25,26).
Unlike cholesterol levels and blood pressure, MI and stroke are not continuously variable, or even quantitative, traits. To explain such discontinuous non-Mendelian traits, the theory of polygenic inheritance was extended by postulating a continuously variable susceptibility and the existence of a threshold beyond which the development of the disease would occur (27). Thus, affected people would have inherited an unfortunate combination of susceptibility genes pushing them beyond the threshold. The relatives of affected people would, on average, also tend to inherit genes that raise susceptibility, explaining the inheritance of the trait. The greater susceptibility of men to early CAD can be explained by a higher threshold in women than in men.
The multigeneration occurrence of angina pectoris and CAD was noted more than 100 years ago (28), and hundreds of subsequent studies have described the familial aggregation of premature CAD and of risk factors such as elevated cholesterol levels (29). For example, in 1966 Slack and Evans (29a) studied first-degree relatives of 121 male and 96 female cases with “premature” CAD (defined as age 55 years or less for men and age 65 years or less for women, the sex difference being due to the fact that men tend to develop CAD earlier than women). Male relatives of male index cases had a fivefold increased incidence, whereas male relatives of female cases had a sevenfold increased incidence of risk. This is consistent with the threshold model because, according to the model, to develop CAD a woman would have to carry more deleterious genes than would a man and therefore she would tend to pass more to her children. Extensive family studies in Finland in the 1970s revealed 3.5-fold increased risk of CAD in brothers of male CAD cases and a 2-fold increased risk in sisters (30,31). In one study of premature CAD in which 19 different risk factors were assessed, the most significant factor was family history (32). A large number of studies have consistently shown a clustering of CAD, with particularly strong familial effects in early-onset disease (1,29,33,34,35,36,37,38).
Of course, families share environments as well as genes, and although there are statistical ways of distinguishing genetic and environmental factors in family studies, it is not straightforward. One approach to the problem is to compare dizygotic and monozygotic twins. These studies are generally consistent with a strong genetic component: monozygotic twins, which share 100% of their genes, exhibit much higher concordance rates for CAD and stroke than do dizygotic twins, which share on average 50% of their genes. For example, a recent study in Sweden examined 21,004 twins. Among men, the relative hazard of death from CAD when one’s twin died of CAD before the age of 55 years was 8.1 for monozygotic twins and 3.8 for dizygotic twins. Among women, when the twin died of CAD before the age of 65 years, the relative hazard was 15.0 for monozygotic twins and 2.6 for dizygotic twins (39).
Overall estimates of the heritability of MI, the major clinical outcome of atherosclerosis, have varied widely, ranging from very little to greater than 50% (39). It is important to note that heritability varies from population to population. In populations in which the environment is relatively homogeneous, say in Iceland, genetics will tend to predominate, whereas in populations exhibiting large environmental differences, say, in Los Angeles, genetics will be relatively less important. Heritability is also a function of the age of the individuals under study, and early-onset forms of CAD tend to have a much stronger genetic component. For example, at older ages the monozygotic/dizygotic relative risk ratio approaches 1 (29). The known risk factors for atherosclerosis generally exhibit very significant heritability estimates as well (Table 95.2). The
strongest evidence for a genetic component is the identification of DNA variations that segregate with the disease in families or associate with the disease in populations. As discussed later, there are several clear examples of such variations for CAD.
strongest evidence for a genetic component is the identification of DNA variations that segregate with the disease in families or associate with the disease in populations. As discussed later, there are several clear examples of such variations for CAD.
TABLE 95.2 Genetic and Environmental Risk Factors for Cardiovascular Disease | ||
|---|---|---|
|
An important and still unresolved question is: How much of the genetic component of CAD do known risk factors such as hyperlipidemia, diabetes, and hypertension explain? The answer (which will be partly dependent on both the particular population and environment) has important clinical implications because if most of CAD can be explained by these measurable phenotypes, there will be little room for the development of independent DNA diagnostics. Some epidemiologic studies have suggested that almost all of the CAD familial aggregation be explained by conventional risk factors (30,31), whereas others have found that the genetic component in CAD requires additional environmental or genetic factors (29,32). The genetic epidemiologic and experimental studies discussed later are consistent with the latter conclusion.
Common Forms of Atherosclerosis Have an Important Environmental Component
The importance of environment in CAD has been dramatically demonstrated in population migration studies. For example, Japanese have a much lower incidence of CAD than Americans, but Japanese-Americans who have adopted a “Western” lifestyle have about the same incidence as other Americans. One study compared Japanese living in Japan, Hawaii, and the mainland United States. Fat consumption increased progressively in the three groups, as did levels of serum cholesterol and CAD (40,41). A dramatic example of a stationary population that has undergone a change of environment resulting in increased disease frequency is the Pima Indians of Arizona. Before adopting a Western lifestyle, these Indians had a modest incidence of obesity and diabetes, but at present the disorders are rampant (42). The influence of environmental factors on lipoprotein metabolism and CAD has also been dramatically revealed by comparisons of different populations. The Seven Countries Study (43) demonstrated strong positive correlations among the consumption of saturated fats, serum cholesterol levels, and CAD mortality among different populations. From these and other studies, it appears that the difference in CAD incidence among populations is due primarily to environmental factors. This is not to say, however, that populations do not differ in genetic susceptibility; for example, the Pima Indians have a surprisingly low incidence of CAD given the high frequency of diabetes.
A very interesting and potentially important environmental factor in atherosclerosis and metabolic syndrome is related to the pathologic or nutritional status of the mother during fetal development (44). Numerous human and experimental studies have revealed that intrauterine undernutrition is associated with obesity and other detrimental metabolic characteristics in adulthood. Using a mouse model, Yura et al. (45) observed that offspring with fetal undernutrition exhibited a premature onset of neonatal leptin surge compared to offspring with normal nutrition. This leptin surge appears to be involved in the formation of the energy-regulation circuits in the hypothalamus that affect metabolism in adults. Perhaps related to this phenomenon, the observation that maternal hypercholesterolemia is associated with increased progression of atherosclerosis has recently been experimentally confirmed in rabbits and mice (46).
An important but poorly understood aspect of the genetics of atherosclerosis involves the role of genetic variation in the response to dietary or other environmental changes. It is well known, for example, that different individuals respond very differently to challenge with a high-fat diet. and some striking ethnic differences in dietary responses have been documented (47,48). In some instances, specific genetic factors have been shown to contribute to genetic–environmental interactions. For example, a common polymorphism of the fibrinogen gene appears to influence MI primarily in individuals who smoke (49,50). In addition, a common polymorphism of MTHFR, an enzyme in the folate cycle, causes high levels of homocysteine if the intake of folate is low (51,52). Genetic–environmental interactions are very difficult to study in detail in humans. One problem relates to the inability to control the environment carefully, but the major problem is that a genotype cannot be replicated in humans (except in identical twins). Therefore, it is almost impossible to examine the effect of a number of different environments on a phenotype while holding the genotype constant. Consequently, the data available for humans represent average results over many different genotypes. An excellent potential model for detailed studies of genetic–environmental interactions is the mouse because many different inbred strains, each representing a unique gene pool, have been derived by brother–sister mating. Numerous differences in dietary responsiveness have been observed among strains of mice, affecting traits such as plasma lipoproteins and lesion development (53,54). For example, a locus on mouse chromosome 2 determines the effect of a high-fat, high-cholesterol diet on the change in plasma cholesterol levels (54). Sing and colleagues provide a detailed discussion of the complexities of gene–environment interactions in atherosclerosis (55).
Genetic Dissection of Atherosclerosis
Our understanding of the genes underlying Mendelian traits has been revolutionized by positional cloning, a method for disease gene identification that does not depend on an understanding of the biochemistry of the trait. Although the genes for certain disorders such as FH were identified by functional cloning (first identifying the protein that underlies the disease and then the gene), most of the genes in Table 95.1 were discovered by first mapping the disease gene in families, then examining positional candidates in the region for mutations that occur only in affected individuals, and lastly examining the protein product. In the case of Mendelian traits, it is usually straightforward to construct a transmission model to test whether the disease gene segregates with genetic markers (Fig. 95.2). The model includes parameters such as the mode of inheritance (e.g., recessive, dominant, or X-linked), the recombination fraction between the gene and the genetic markers, and, in some cases, nongenetic contributions. The evidence for linkage is usually expressed as a “log of the odds” or “lod” score. This is the base-10 logarithm of the odds ratio, which is the probability of the data given linkage divided by the probability of the data in the absence of linkage. In the case of human studies, an odds ratio of 1,000, equivalent to a lod score of 3.0, is considered strong evidence (p < .05) of linkage. Such a high odds ratio is necessary because in linkage analysis many comparisons (the trait vs. each genetic marker) are performed.
Generally, the construction of a transmission model is not feasible for complex traits in which many genetic and environmental factors contribute to the phenotype. Although mathematical modeling approaches such as complex segregation analysis have provided information about the existence of “major genes” with large effects on a trait, it is very difficult to
construct realistic transmission models due to unknowns such as the number of genes involved, the number of alleles of each gene and their frequencies, the impact of each allele on a trait, and gene–environment and gene–gene interactions. Thus, progress in identifying genes for common forms of atherosclerosis has been slow.
construct realistic transmission models due to unknowns such as the number of genes involved, the number of alleles of each gene and their frequencies, the impact of each allele on a trait, and gene–environment and gene–gene interactions. Thus, progress in identifying genes for common forms of atherosclerosis has been slow.
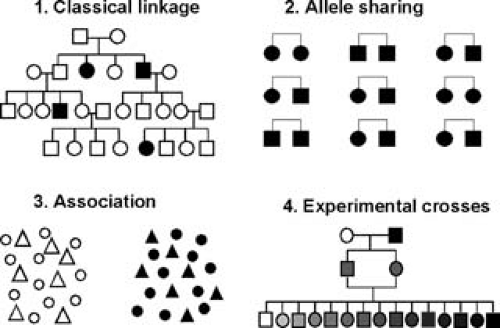 FIGURE 95.2. Four approaches for the dissection of complex genetic traits. Classical linkage analysis (1) involves the construction of a model for the segregation of genes that contribute to the trait. Such models are relatively straightforward to construct for Mendelian traits but are difficult to devise for most multifactorial traits. Allele-sharing methods (2) provide a model-free approach for linkage analysis of complex traits. Usually, pairs of siblings are studied, although the method can be extended to various relations. The basic concept of the method is that affected pairs tend to have increased sharing of chromosomal regions that harbor disease genes (as recognized by genetic markers). Association analysis (3) involves testing whether particular polymorphisms occur more frequently in affected individuals than in controls. The last approach involves the use of experimental crosses with animal models (4). Rats and mice are the most useful mammals for genetic studies, and different strains of these rodents exhibit many variations relevant to atherosclerosis, including differences in susceptibility to arterial lesions in the presence of hyperlipidemia and variations in lipid metabolism, blood pressure, obesity, insulin resistance, and artery wall functions. Genetic crosses of such strains can produce hundreds of unique progeny that inherit different parental alleles contributing to the traits, and linkage analysis is more powerful than with human studies of comparable size. This figure is patterned after figures in a review by Lander and Schork (181). |
Genetic contributions to common, complex forms of atherosclerosis (and of traits relevant to the disease) were first studied by population association studies with “candidate genes” (genes chosen based on their biologic functions) (Fig. 95.3). One of the first important examples of this was apolipoprotein E (apoE), which was found to exhibit three common alleles in all populations studied (E2, E3, E4). The E2 form was found to be strongly associated with low cholesterol levels in the population, and the E4 allele was associated with elevated cholesterol levels (56). The basis of this effect is due in part to differences in the ability of the apoE isoforms to bind to the LDL receptor and the LDL receptor–related protein (LRP). Interestingly, homozygosity for the E2 allele is also strongly associated with the relatively uncommon dyslipidemia type III; because E2 is usually associated with decreased cholesterol levels, type III presumably involves other genetic or environmental factors. Since the early 1980s, thousands of association studies with candidate genes have been performed for traits relevant to atherosclerosis. Of these, some are convincing (Table 95.3), but most remain questionable, including many that have been studied in multiple, relatively large populations (57). One of the problems with association studies is the possible confounding effect of population admixture. Thus, in heterogeneous populations consisting of multiple ethnic groups, spurious associations are likely to occur because the groups generally differ in allele frequencies and might also differ in disease susceptibilities. This problem can be addressed by techniques such as the transmission disequilibrium test involving family-based associations [e.g., (58)], but such tests have rarely been used because they require large numbers of families.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


