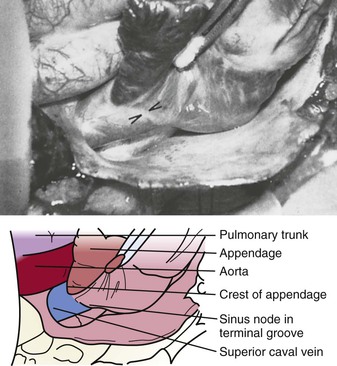
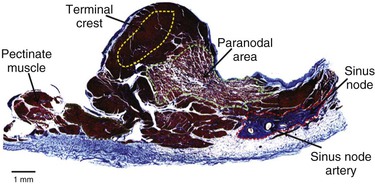
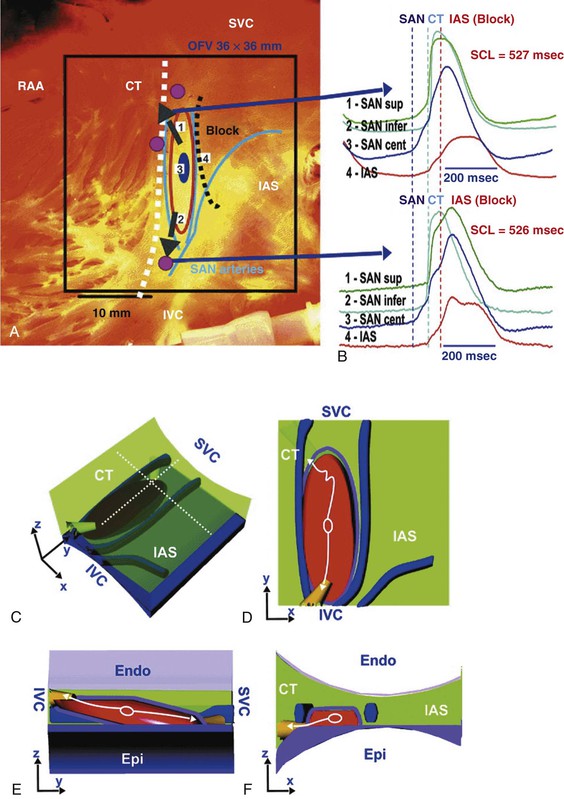
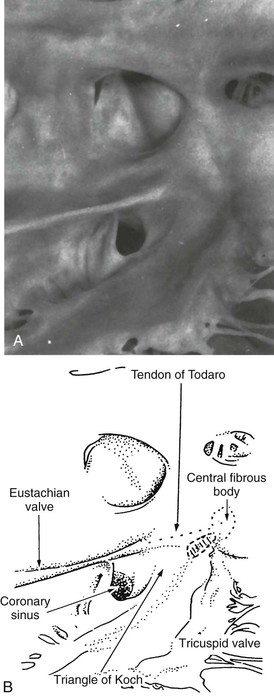
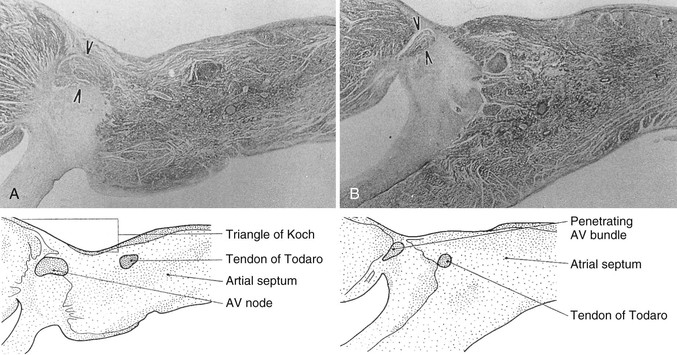
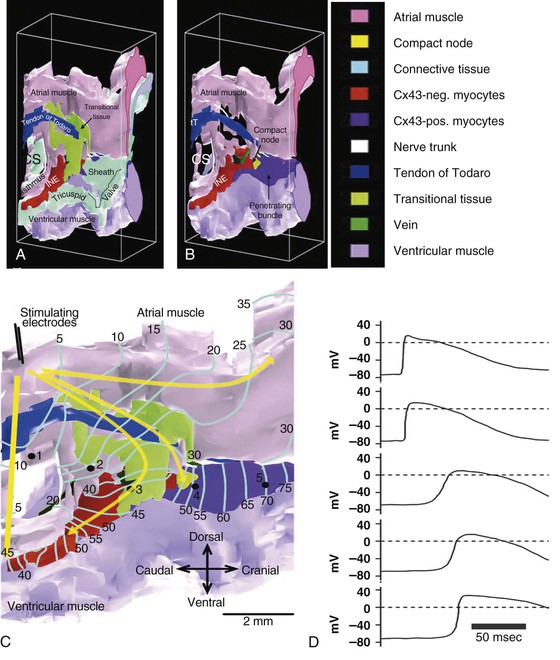
Michael Rubart, Douglas P. Zipes In humans, the sinoatrial node is a spindle-shaped structure composed of a fibrous tissue matrix with closely packed cells. It is 10 to 20 mm long and 2 to 3 mm wide and thick and tends to narrow caudally toward the inferior vena cava. It lies less than 1 mm from the epicardial surface, laterally in the right atrial sulcus terminalis at the junction of the superior vena cava and right atrium (Figs. 33-1 and 33-2). The artery supplying the sinoatrial node branches from the right (55% to 60% of the time) or the left (40% to 45%) circumflex coronary artery and approaches the node from a clockwise or counterclockwise direction around the junction of the superior vena cava and right atrium. Based on histology and immunolabeling, the normal AV junctional area (Figs. 33-5 and 33-6) is composed of multiple distinct structures, including transitional tissue, inferior nodal extension, compact portion, penetrating bundle, His bundle, atrial and ventricular muscle, central fibrous body, tendon of Todaro, and valves.10,11 Figure 33-7A, B shows a computer-generated three-dimensional reconstruction of the AV junctional area in a rabbit heart. At the level of the AV junction, the tract of nodal tissue is divided into two major components, the inferior nodal extension and the penetrating bundle (red and purple areas, respectively, in Fig. 33-7A, B). The inferior nodal extension is located between the coronary sinus and the tricuspid valve, and the end of the inferior nodal extension is covered by transitional tissue (light green area in Fig. 33-7A, B). The small myocytes in the inferior nodal extension are dispersed among connective tissue and do not express connexin 43, whereas myocytes in the transitional zone do express connexin 43; however, unlike the connexin 43–positive atrial myocytes in the working myocardium, they are loosely packed between collagen septa. The inferior nodal extension is continuous with the penetrating bundle, which penetrates the fibrous tissue separating the atria and ventricles and emerges in the ventricles as the bundle of His. Both structures are covered by connective tissue (sheaths in Fig. 33-7A) and are therefore enclosed. Myocytes in the penetrating bundle express connexin 43 and are dispersed among connective tissue. A tract of connexin 43–positive nodal tissue projects into the connexin 43–negative inferior nodal extension. The compact portion of the AV node (yellow area in Fig. 33-7A, B) is a superficial structure lying just beneath the right atrial endocardium, anterior to the ostium of the coronary sinus, and directly above the insertion of the septal leaflet of the tricuspid valve. It is at the apex of a triangle formed by the tricuspid annulus and the tendon of Todaro (blue area in Fig. 33-7A, B), which originates in the central fibrous body and passes posteriorly through the atrial septum to continue with the eustachian valve (see Figs. 33-5 and 33-6A). The term triangle of Koch, however, has to be used with caution because histologic studies of anatomically normal adult hearts have demonstrated that the tendon of Todaro, which forms one side of the triangle of Koch, is absent in about two thirds of hearts. The compact node is located at the junction where the connexin 43–negative nodal tissue (red area in Fig. 33-7A, B) meets the connexin 43–positive nodal tissue (purple area in Fig. 33-7A, B). Myocytes in the nodal portion are small and weakly positive for connexin 43. In 85% to 90% of human hearts, the arterial supply to the AV node is derived from a branch of the right coronary artery that originates at the posterior intersection of the AV and interventricular grooves (crux). A branch of the circumflex coronary artery provides the arterial supply to the AV node in the remaining hearts. Fibers in the lower part of the AV node may exhibit automatic impulse formation.11 The main function of the AV node is to delay transmission of atrial impulses to the ventricles, thereby coordinating atrial and ventricular contractions (Fig. 33-7C, D). During normal anterograde AV conduction, the action potential propagates from the sinoatrial node through atrial working myocardium (the existence of specialized internodal conduction pathways has been controversial) and enters the tract of nodal tissue at two points (see Fig. 33-7C; see also Video 33-1 VIDEO 33-1 Simulation of anterograde conduction through the atrioventricular node (AVN) using an electroanatomical model. The preparation is stimulated at the interatrial septum as shown by the stimulating electrodes. There is a flash and click coincident with the stimulus. (From Li J, Greener ID, Inada S, et al: Computer three dimensional reconstruction of the atrioventricular node. Circ Res 102:975, 2008.) This structure is the continuation of the penetrating bundle on the ventricular side of the AV junction before it divides to form the left and right bundles (see Fig. 33-6A). Myocytes in the His bundle are small and connexin 43 positive (see Fig. 33-7C). However, large, well-formed fasciculoventricular connections between the penetrating portion of the AV bundle and the ventricular septal crest are rarely found in adult hearts. Branches from the anterior and posterior descending coronary arteries supply the upper muscular interventricular septum with blood, which makes the conduction system at this site more impervious to ischemic damage unless the ischemia is extensive. These structures begin at the superior margin of the muscular interventricular septum, immediately beneath the membranous septum, with cells of the left bundle branch cascading downward as a continuous sheet onto the septum beneath the noncoronary aortic cusp (Fig. 33-8A). The AV bundle may then give off other left bundle branches, sometimes constituting a true bifascicular system with an anterosuperior branch, in other hearts giving rise to a group of central fibers, and in still others appearing more as a network without clear division into a fascicular system (Fig. 33-8B). The right bundle branch continues intramyocardially as an unbranched extension of the AV bundle down the right side of the interventricular septum to the apex of the right ventricle and base of the anterior papillary muscle. In some human hearts, the His bundle traverses the right interventricular crest and gives rise to a right-sided narrow stem origin of the left bundle branch. The anatomy of the left bundle branch system can be variable and not conform to a constant bifascicular division. However, the concept of a trifascicular system remains useful to both electrocardiographers and clinicians (see Chapter 12). These fibers connect with the ends of the bundle branches to form interweaving networks on the endocardial surface of both ventricles and transmit the cardiac impulse almost simultaneously to the entire right and left ventricular endocardium. Purkinje fibers tend to be less concentrated at the base of the ventricle and at the papillary muscle tips. They penetrate the myocardium for varying distances, depending on the animal species. In humans, they apparently penetrate only the inner third of the endocardium, whereas in pigs, they almost reach the epicardium. Such variations could influence changes produced by myocardial ischemia, for example, because Purkinje fibers appear to be more resistant to ischemia than ordinary myocardial fibers are. Purkinje myocytes are found in the His bundle and bundle branches, cover much of the endocardium of both ventricles (see Fig. 33-8B), and align to form multicellular bundles in longitudinal strands separated by collagen. Although conduction of cardiac impulses appears to be their major function, free-running Purkinje fibers composed of many Purkinje cells in a series, sometimes called false tendons, are capable of contraction. Action potentials propagate within the thin Purkinje fiber bundles from the base to the apex before activation of the surrounding myocytes occurs. Purkinje myocytes largely lack transverse tubules (Fig. e33-1 Pathways of Innervation. The AV node and His bundle region are innervated by a rich supply of cholinergic and adrenergic fibers with densities exceeding those found in the ventricular myocardium.14 Immunolabeling with markers for sympathetic and parasympathetic nerves revealed nonuniform innervation density in the AV junctional area. For example, the inferior nodal extension has been shown to exhibit a higher density of both nerve types than the working atrial myocardium does, whereas the opposite is true for the compact node.15 Ganglia, nerve fibers, and nerve nets lie close to the AV node. Parasympathetic nerves to the AV node region enter the canine heart at the junction of the inferior vena cava and the inferior aspect of the left atrium, adjacent to the entrance to the coronary sinus. Nerves in direct contact with AV nodal fibers have been noted, along with agranular and granular vesicular processes, which presumably represent cholinergic and adrenergic processes. In general, autonomic neural input to the heart exhibits some degree of “sidedness,” with the right sympathetic and vagal nerves affecting the sinoatrial node more than the AV node and the left sympathetic and vagal nerves affecting the AV node more than the sinoatrial node. The distribution of neural input to the sinoatrial and AV nodes is complex because of substantial overlapping innervation. Despite the overlap, specific branches of the vagal and sympathetic nerves can be shown to innervate certain regions preferentially. Supersensitivity to acetylcholine follows vagal denervation. Stimulation of the right stellate ganglion produces sinus tachycardia with less effect on AV nodal conduction, whereas stimulation of the left stellate ganglion generally produces a shift in the sinus pacemaker to an ectopic site and consistently shortens AV nodal conduction time and refractoriness but inconsistently speeds the sinoatrial nodal discharge rate. Stimulation of the right cervical vagus nerve primarily slows the sinoatrial nodal discharge rate, and stimulation of the left vagus primarily prolongs AV nodal conduction time and refractoriness when sidedness is present. Neither sympathetic nor vagal stimulation affects normal conduction in the His bundle. The negative dromotropic response of the heart to vagal stimulation is mediated by the activation of IK.Ach,Ade, which results in hyperpolarization of the AV nodal cells and thereby influences the conductive properties of the node. The positive dromotropic effect of sympathetic stimulation arises as a consequence of an increase in cytosolic cAMP levels and ensuing activation of the L-type Ca2+ current ICa.L (see Table 33-3). Most efferent sympathetic impulses reach the canine ventricles over the ansae subclaviae, branches from the stellate ganglia. Sympathetic nerves then synapse primarily in the caudal cervical ganglia and form individual cardiac nerves that innervate relatively localized parts of the ventricles. The major route to the heart is the recurrent cardiac nerve on the right side and the ventrolateral cardiac nerve on the left. In general, the right sympathetic chain shortens refractoriness primarily of the anterior portion of the ventricles, and the left affects primarily the posterior surface of the ventricles, although overlapping areas of distribution occur. The intraventricular route of sympathetic nerves generally follows the coronary arteries. Functional data have suggested that afferent and efferent sympathetic nerves travel in the superficial layers of the epicardium and dive to innervate the endocardium, and anatomic observations have supported this conclusion. Vagal fibers travel intramurally or subendocardially and rise to the epicardium at the AV groove (Fig. 33-9A). Sympathetic nerve density in the left ventricle appears to be higher in the epicardial than in the endocardial portion of the ventricle, which at least in part results from transmural gradients in the expression of cytokines during cardiac development that attract and repel, respectively, sympathetic nerve growth (Fig. 33-9B).14,16 Effects of Vagal Stimulation. The vagus modulates cardiac sympathetic activity at prejunctional and postjunctional sites by regulating the amount of norepinephrine released and by inhibiting cAMP-induced phosphorylation of cardiac proteins, including ion channels and calcium pumps. The latter inhibition occurs at more than one level in the series of reactions constituting the adenylate cyclase–, cAMP-dependent protein kinase system. Neuropeptides released from the nerve fibers of both autonomic limbs also modulate autonomic responses. For example, NPY released from sympathetic nerve terminals inhibits cardiac vagal effects. Tonic vagal stimulation produces a greater absolute reduction in the sinoatrial rate in the presence of tonic background sympathetic stimulation, a sympathetic-parasympathetic interaction termed accentuated antagonism. In contrast, changes in AV conduction during concomitant sympathetic and vagal stimulation are essentially the algebraic sum of the individual AV conduction responses to tonic vagal and sympathetic stimulation alone. Cardiac responses to brief vagal bursts begin after a short latency and dissipate quickly; in contrast, cardiac responses to sympathetic stimulation commence and dissipate slowly. The rapid onset and offset of responses to vagal stimulation allow dynamic beat-to-beat vagal modulation of the heart rate and AV conduction, whereas the slow temporal response to sympathetic stimulation precludes any beat-to-beat regulation by sympathetic activity. Periodic vagal bursting, as may occur each time that a systolic pressure wave arrives at the baroreceptor regions in the aortic and carotid sinuses, induces phasic changes in sinus cycle length and can entrain the sinus node to discharge faster or slower at periods identical to those of the vagal burst. In a similar phasic manner, vagal bursts prolong AV nodal conduction time and are influenced by background levels of sympathetic tone. Because the peak vagal effects on sinus rate and AV nodal conduction occur at different times in the cardiac cycle, a brief vagal burst can slow the sinus rate without affecting AV nodal conduction or can prolong AV nodal conduction time and not slow the sinus rate. Bilateral but not unilateral vagal nerve stimulation increases and reverses the spatial dispersion of ventricular repolarization as the direction of repolarization from the apex to the base in sinus rhythm shifts from the base to the apex. This effect is attributable to more pronounced prolongation of the action potential at the apex than at the base of the heart (Fig. e33-2 Effects of Sympathetic Stimulation. Similar to bilateral vagal nerve stimulation, sympathetic nerve stimulation also increases and reverses the spatial gradients of ventricular repolarization as the direction of polarization from the apex to the base in sinus rhythm shifts from the base to the apex. This reversal results from a marked shortening of action potential duration at the base, with no or very little effect on the repolarization time course at the apex of the heart (see Fig. e33-2).17 Nonuniform distribution of sympathetic nerves—and thus norepinephrine levels—may in part contribute to some of the nonuniform electrophysiologic effects because the ventricular content of norepinephrine is greater at the base than at the apex of the heart.11 In humans, both direct and reflex sympathetic stimulation increases regional differences in cardiac repolarization. The dispersion of repolarization is significantly enhanced in patients with ischemic cardiomyopathy.18 Afferent vagal activity appears to be higher in the posterior ventricular myocardium, which may account for the vagomimetic effects of inferior myocardial infarction. The vagi exert minimal but measurable effects on ventricular tissue; they decrease the strength of myocardial contraction and prolong refractoriness. Under some circumstances, acetylcholine can cause a positive inotropic effect. It is now clear that the vagus (acetylcholine) can exert direct effects on some types of ventricular fibers, as well as indirect effects by modulating sympathetic influences. Beyond the beat-to-beat regulation of rate and contractile force, sympathetic input to the heart, through both translational and post-translational modifications, also exerts long-term regulation of adrenergic receptor sensitivity and ionic channels. These long-term changes in autonomic responsiveness and cardiac electrical properties appear to be mediated, at least in part, by highly localized signaling cascades involving neurally released molecules such as NPY.19 Alterations in vagal and sympathetic innervation (autonomic remodeling) can influence the development of arrhythmias and result in sudden cardiac death from ventricular tachyarrhythmias.20 Damage to nerves extrinsic to the heart, such as the stellate ganglia, and to intrinsic cardiac nerves from diseases that may affect primarily nerves, such as viral infections, or from diseases that secondarily cause cardiac damage may produce cardioneuropathy. Although the mechanisms by which altered sympathetic innervation modulates cardiac electrical properties are largely unknown, spatially heterogeneous sympathetic hyperinnervation could result in enhanced dispersion of myocardial excitability and refractoriness via patchy adrenergic stimulation of ionic currents, including ICa.L, IKs, and ICl (see Table 33-3). Sympathetic hypoinnervation has been shown to increase the sensitivity of adrenergic receptors to activation by circulating catecholamines (denervation supersensitivity).14 Numerous studies have suggested a primary role of altered cardiac sympathetic innervation in arrhythmogenesis. Chronic infusion of nerve growth factor into the left stellate ganglion in dogs with chronic myocardial infarction and complete AV block caused spatially heterogeneous sympathetic cardiac hyperinnervation (nerve sprouting) and dramatically increased the incidence of sudden death from ventricular tachyarrhythmias.20 Ambulatory long-term recordings of left stellate ganglion nerve activity in these dogs revealed that most malignant ventricular arrhythmias were preceded by increased neuronal discharge, thus suggesting a causal role of sympathetic input in triggering arrhythmogenic sudden cardiac death.21 A high-cholesterol diet was reported to result in cardiac sympathetic hyperinnervation in rabbits and a marked increase in the incidence of ventricular fibrillation (VF).22 Explanted human hearts from transplant recipients with a history of arrhythmias exhibited a significantly higher and also more heterogeneous density of sympathetic nerve fibers than did those from patients without arrhythmias (Fig. 33-10A). Whether neural remodeling also involved parasympathetic nerve fibers in the heart was not examined in these studies. In patients with congestive heart failure, sympathetic neural tone is upregulated, and excess activation of the sympathetic nervous system leads to adverse myocardial effects, including lethal arrhythmias, and also causes depletion of cardiac norepinephrine content. This depletion of norepinephrine has recently been shown to result, at least partially, from neurotransmitter switching and transdifferentiation from catecholaminergic into cholinergic neurons in the chronically failing heart (Fig. 33-10B).23 This process is induced by release of cholinergic differentiation factors from failing cardiomyocytes. It remains to be determined, however, whether neurotransmitter switching is an adaptive response to protect the heart from excess sympathetic stimulation and thus lethal arrhythmias. Interestingly, beta adrenoceptor blockade in rats with coronary artery ligation reversed the myocardial sympathetic axon depletion in intact myocardium remote from the infarct but did not affect peri-infarct sympathetic hyperinnervation.24
Genesis of Cardiac Arrhythmias
Electrophysiologic Considerations
Anatomy of the Cardiac Conduction System
Sinoatrial Node
Atrioventricular Junctional Area and Intraventricular Conduction System
Atrioventricular Node
 ). The first point is at the end of the inferior nodal extension (next to the penetrating bundle) via the transitional tissue. This conduction pathway most likely corresponds to the fast-pathway route previously observed in electrical mapping experiments.11 Second, the action potential enters toward the beginning of the inferior nodal extension. This conduction pathway probably constitutes the slow-pathway route. The action potential cannot enter the nodal tissue at other tissue points because the nodal and atrial tissues are isolated from each other by a vein along this length of tissue (dark green area in Fig. 33-7B, C). From the two entry points, the action potentials propagate both anterogradely and retrogradely along the inferior nodal extension and eventually annihilate each other. The action potential entering the nodal tract via the transitional zone also propagates into the compact node and then reaches the His bundle and propagates down the left and right bundle branches. Transmembrane action potentials recorded from in situ cardiomyocytes at various locations within the nodal tract exhibit distinct shapes and time courses (see Fig. 33-7D). Action potentials from extranodal atrial tissue and the His bundle (locations 1 and 5, respectively, in Fig. 33-7C) have more hyperpolarized diastolic potentials and faster upstrokes than do myocytes in the transitional zone (location 3) and penetrating bundle (location 4). This smaller rate of depolarization results in slowing of conduction across the compact portion and penetrating bundle (conduction velocity, <10 cm/sec versus 35 cm/sec in atrial working myocardium), thereby giving rise to the AV conduction delay.
). The first point is at the end of the inferior nodal extension (next to the penetrating bundle) via the transitional tissue. This conduction pathway most likely corresponds to the fast-pathway route previously observed in electrical mapping experiments.11 Second, the action potential enters toward the beginning of the inferior nodal extension. This conduction pathway probably constitutes the slow-pathway route. The action potential cannot enter the nodal tissue at other tissue points because the nodal and atrial tissues are isolated from each other by a vein along this length of tissue (dark green area in Fig. 33-7B, C). From the two entry points, the action potentials propagate both anterogradely and retrogradely along the inferior nodal extension and eventually annihilate each other. The action potential entering the nodal tract via the transitional zone also propagates into the compact node and then reaches the His bundle and propagates down the left and right bundle branches. Transmembrane action potentials recorded from in situ cardiomyocytes at various locations within the nodal tract exhibit distinct shapes and time courses (see Fig. 33-7D). Action potentials from extranodal atrial tissue and the His bundle (locations 1 and 5, respectively, in Fig. 33-7C) have more hyperpolarized diastolic potentials and faster upstrokes than do myocytes in the transitional zone (location 3) and penetrating bundle (location 4). This smaller rate of depolarization results in slowing of conduction across the compact portion and penetrating bundle (conduction velocity, <10 cm/sec versus 35 cm/sec in atrial working myocardium), thereby giving rise to the AV conduction delay.
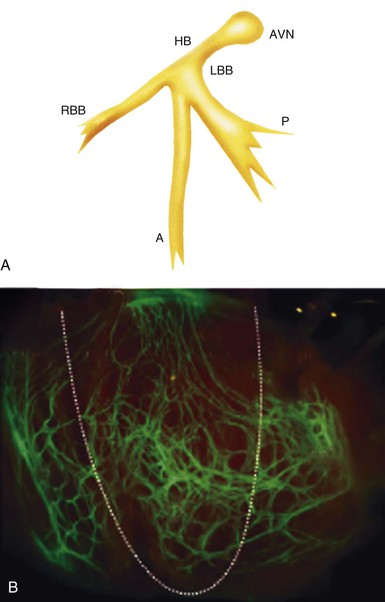
Bundle of His (Penetrating Portion of the Atrioventricular Bundle)
Bundle Branches (Branching Portion of the Atrioventricular Bundle)
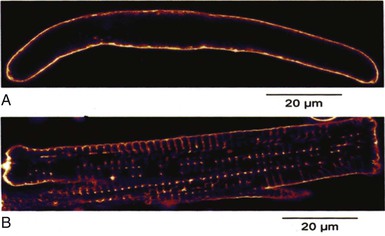
Terminal Purkinje Fibers
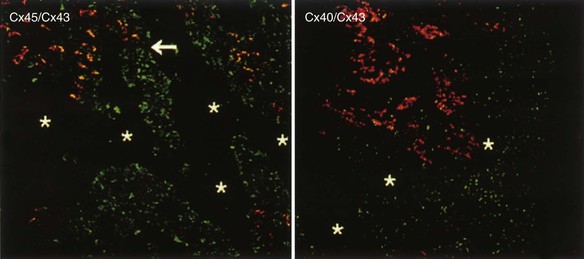
 ), which reduces membrane capacitance and thus accelerates action potential propagation.12 Propagation of action potentials within the His-Purkinje system and working myocardium is mediated by connexins. Ventricular myocytes express mainly connexin 43, and Purkinje fibers express connexins 40 and 45. The molecular identity of the connexin type that enables transmission of impulses at the Purkinje fiber–myocyte junction (PMJ) is unclear. It is also still not clear how the small amount of depolarizing current provided by the thin bundle of Purkinje fibers can activate a much larger mass of ventricular muscle (current-to-load mismatch).13 It is possible that individual gap junctional channels at the PMJ are formed by more than one connexin isoform. These disparate connexin phenotypes may create specific types of hybrid channels with unique properties that ensure safe conduction at the PMJ. Because Purkinje cells have markedly longer repolarization times than surrounding myocytes do (see Fig. 33-17E), these connexin hybrids could also decrease entrainment of repolarization at the PMJ and thereby increase repolarization gradients.
), which reduces membrane capacitance and thus accelerates action potential propagation.12 Propagation of action potentials within the His-Purkinje system and working myocardium is mediated by connexins. Ventricular myocytes express mainly connexin 43, and Purkinje fibers express connexins 40 and 45. The molecular identity of the connexin type that enables transmission of impulses at the Purkinje fiber–myocyte junction (PMJ) is unclear. It is also still not clear how the small amount of depolarizing current provided by the thin bundle of Purkinje fibers can activate a much larger mass of ventricular muscle (current-to-load mismatch).13 It is possible that individual gap junctional channels at the PMJ are formed by more than one connexin isoform. These disparate connexin phenotypes may create specific types of hybrid channels with unique properties that ensure safe conduction at the PMJ. Because Purkinje cells have markedly longer repolarization times than surrounding myocytes do (see Fig. 33-17E), these connexin hybrids could also decrease entrainment of repolarization at the PMJ and thereby increase repolarization gradients.
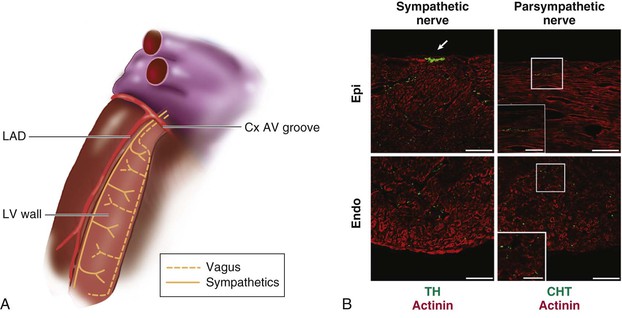
Innervation of the Atrioventricular Node, His Bundle, and Ventricular Myocardium
 ).17
).17
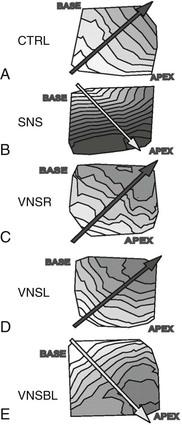
Arrhythmias and the Autonomic Nervous System
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Genesis of Cardiac Arrhythmias
33