Cystic fibrosis transmembrane conductance regulator (CFTR) gene and protein
From the functional aspect, mutations are grouped in five classes (I–V). Most of these cause complete channel failure, whether they affect the biosynthesis of the protein (class I), its maturity process (class II), or its function (class III). In contrast, in class IV and V mutations, the proteins produced retain certain residual activity. Generally, class I, II, and III mutations are related to serious manifestations of the disease, which include pancreatic insufficiency, whereas class V mutations present mild manifestations, or even oligo-symptomatic (traditionally named nonclassic presentations, and currently named diseases related to CTFR), such as the congenital bilateral absence of the vas deferens (CBAVD), idiopathic chronic pancreatitis, and bronchiectasis, among others.
Understanding of the physiopathological nature of the disease at the molecular level has improved during the past years. This knowledge prompted the development of new therapeutic strategies oriented to correct the gene dysfunction. Thus, genotyping patients with CF also became important for determining specific treatments according to the mutation class (allele-specific therapies).
Diagnosis
The disease is diagnosed using clinical criteria and laboratory methods. The presence of two or more of the following clinical criteria suggest the disease: chronic pulmonary disease, chronic sinus disease, malabsorption, obstructive azoospermia in males, family history of cystic fibrosis, and salt-wasting syndrome.
Two positive sweat tests
Presence of two mutations in the CFTR gene
An abnormal transepithelial membrane potential
Important clinical elements for diagnosis
Respiratory |
Upper airway |
Nasal polyps |
Chronic sinusitis |
Lower airway |
Persistent tachypnea and retraction |
Wheezing with persistent hyperinflation |
Chronic cough |
Sputum culture positive for Haemophilus influenzae, Staphylococcus aureus, or Pseudomonas aeruginosa |
Recurrent pneumonia |
Bronchiectasis |
Hemoptysis |
Chest X-rays |
Bilateral air trapping |
Persistent atelectasis (particularly in the right upper lobe) |
Gastrointestinal |
Meconium ileus |
Prolonged neonatal jaundice |
Malabsorption–steatorrhea |
Rectal prolapse |
Cirrhosis and portal hypertension |
Other |
Growth failure |
Positive family history |
Salty sweat |
Edema and hyponatremia |
Digital clubbing |
Azoospermia and absence of vas deferens |
Metabolic alkalosis |
- (a)
Poor knowledge about the disease among health professionals
- (b)
Lack of information for timely diagnosis within the first health attention levels
- (c)
Diagnosis inaccessibility, given difficulties to conduct the sweat test at a location near the patient’s domicile
Clinical Manifestations
Lung Compromise
The onset age of respiratory symptoms is variable, and some symptoms can appear even during the neonatal phase, although they always appear during the first years of life. The symptoms may be nonspecific, but the pediatrician must suspect and consider CF.
Respiratory disease causes greater morbidity and mortality rates. More than 95% of the patients with CF present with respiratory symptoms, and with malabsorption, it is the most common clinical presentation.
- 1.
Recurrent or prolonged bronchiolitis
- 2.
Recurrent or persistent atelectasis
- 3.
Chronic bronchitis, with cough and mucopurulent expectoration
Older children may present with bronchial obstruction, cough, and expectoration, which vary according to the compromise of the patient. CF must be considered and ruled out for children with asthma symptoms who do not respond to the treatment.
When the disease has progressed because of the persistence of infections and chronic obstruction, bronchiectasis may be observed, with or without hemoptysis.
Extrapulmonary signs of chronic lung disease may be observed, such as increase of chest anteroposterior diameter and digital clubbing.
CF must be ruled out in every child with asthma symptoms who does not respond to treatment with bronchodilators or shows failure to thrive.
A characteristic of the disease, granted by the basic genetic alteration, is infection of the airway, which in many cases ultimately causes a chronic infection. In the first stages of CF, the most frequent agents are nontypeable Haemophilus influenzae (HI) or Staphylococcus aureus (SA). As the disease progresses and the patients become older, Pseudomonas aeruginosa (PA) is the main agent.
The physician should suspect the disease when there is persistence of bronchial secretions in patients who are positive for nontypeable Haemophilus influenzae (HI) or Staphylococcus aureus (SA), and who have not received mechanical ventilation support. If Pseudomonas aeruginosa appears on the cultures, CF must be ruled out.
At first, the chest X-ray may show hyperinflation, but it then progresses to bronchitis with patchy areas with consolidations. These changes are gradual and increase with the progression of the disease.
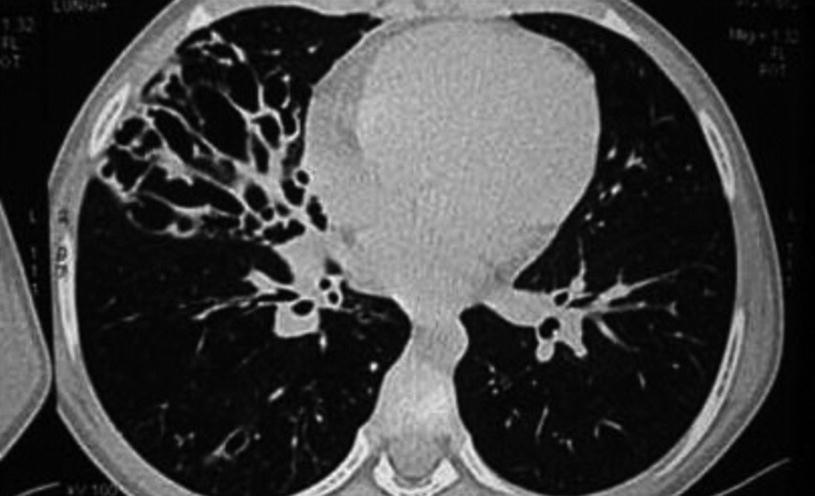
Bronchiectasis. Computerized axial tomography of a 16-year-old patient shows bronchial enlargement and cystic deformations, mainly in the middle lobe
The presence of cylindrical bronchiectasis, mainly in the superior lobes, is suggestive of the disease, which must be ruled out with a sweat test.
Lung compromise is also observed in lung function tests, which can be present even in the early stages of the disease.
Compromise of the upper airway is caused by the hyperreactivity of the mucus secretion glands. This, with the alterations in mucociliary transport, causes hypertrophy and edema in the mucosa membranes, as well as obstruction of sinus ostia.
Almost all patients with cystic fibrosis present with opacification in the paranasal sinus X-rays, and many have chronic sinusitis symptoms, which may cause infectious exacerbations in the lower airway. More than 25% of all patients with CF have nasal polyps.
If there are nasal polyps and the cause has not been established, CF must be ruled out.
Intestinal Obstruction
Meconium ileus appears in 10–15% of patients with CF, and it is the most common manifestation during neonatal age. It occurs secondary to water reduction in the intestines, and most of these patients also have pancreatic insufficiency.
Starting from the 18th week of gestation, it can be recognized through echography. This condition may also appear after childbirth, with abdominal enlargement and scarce stools with mucus, or no defecation at all, as well as biliary vomiting. The obstruction is usually located in the small intestine proximal to the ileocecal valve. Encapsulated meconium peritonitis appears in more than 50% of the patients with meconium ileus.
In the abdominal X-rays, the intestine can appear enlarged, with areas of air mixed with dehydrated meconium, usually in the abdomen right lower quadrant. Calcifications images may also appear.
The presence of meconium ileus in a newborn is almost synonym for CF, and it should be ruled out. The existence of duodenal atresia has also been related to CF in more than 50% of the patients, and therefore any type of intestinal obstruction in an infant is suggestive of CF, which must be ruled out.
Also called distal intestinal obstruction syndrome , this can be an important complication in some patients, particularly in adolescents. It is characterized by constipation, vomiting, abdominal pain, recurrent colic, and palpable fecal matter in the right iliac fossa, or in the right side.
Gastrointestinal Alterations
Neonatal cholestatic jaundice can appear in patients with meconium ileus (50%), but it can also appear in newborns without meconium ileus.
Between 85% and 90% of the patients with cystic fibrosis present with pancreatic insufficiency at birth.
The clinical presentation is abundant fetid stools, with fat presence characteristics (lack of color, shiny, oily). Malabsorption is confirmed using the stool elastase test, or with the collection of 72 h of stools and analyzing the fat contained, named the Van de Kamer test.
About 10% to 15% of the patients have pancreatic sufficiency with some degree of residual pancreatic activity. Given that these patients do not suffer from malabsorption, diagnosis is difficult and it usually takes longer.
Patients with pancreatic sufficiency may develop pancreatic insufficiency as the disease progresses, and therefore elastase must be controlled annually.
In older patients or in adults, persistent or recurrent pancreatitis of unknown cause is suggestive of CF.
It can be present in about 20% of the patients who are under 5 years old and who have never been treated for CF. This sign is secondary to the lack of treatment for intestinal malabsorption.
Around 5% of the patients with CF develop liver disease. The process is characterized by focal or diffuse cirrhosis, and the complications include splenomegaly, varicose veins, and bleeding: this is the cause of death of 1–2% of patients with cystic fibrosis.
Another complication related to liver disease is biliary lithiasis, which has a greater incidence in patients with CF.
Growth Delay
These patients frequently present with a delay in growth, which is caused by a combination of several factors: (a) increase of calorie intake; (b) chronic lung disease; (c) poor digestion, with corresponding intestinal malabsorption; and (d) low appetite, caused by active lung inflammation.
Intestinal malabsorption can be confirmed observing the increase in depositions, with abnormal stool color and consistency. Malabsorption, especially of fats, also involves a deficiency in the absorption of liposoluble vitamins.
Sweat Glands
CFTR causes the sweat glands to be the only ionic channel capable of reabsorbing sweat chloride; therefore, the patient with CF has a chloride concentration in the skin five times greater than that of normal patients, which is almost the same as the plasmatic concentration. This abnormality is the base of the diagnostic test of the disease.
The sweat of the affected patients tends to be salty, and the loss could be so important that salt crystals could appear in the hair line. Salt loss during heat waves can be serious and cause hyponatremic dehydration, as well as serious hypochloremic alkalosis, which require immediate intervention. This loss could be the first presentation of the disease.
Vas Deferens
Even though the testicles of males with CF are normal, the epididymis either cannot be palpated or is reduced, and the vas deferentia are absent in almost every patient.
Up to 95% of males who suffer from CF are infertile because of azoospermia, caused by the congenital bilateral absence of a vas deferens. Women may be sterile because of thickening of the cervical mucus or nutritional state.
Diabetes Mellitus
Changes in the glucose mechanism of CF patients were described in 1938, but the relationship between cystic fibrosis and diabetes was described later, in 1953. The cause of diabetes is the progressive fibrosis of the pancreas, which in principle is only present in the exocrine pancreas and then extends to the endocrine portion of the gland.
Reported incidence in different studies is quite variable, with a range between 21% and 75%. The range is so wide that it may be caused by dissimilar criteria used for different studies.
Edematous Ascitic Syndrome
In children under 6 months of age, about 5% of CF patients may also present edema, anemia, and hypoproteinemia. These patients need to be nutritionally recovered before conducting the sweat test, to avoid the possibility of false-negatives.
The triad edema, anemia, and hyponatremia is considered to be a CF until it been ruled out.
Family History of CF
A family history of CF it is very useful when CT is suspected.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


