Chapter 56 Exocrine Pancreas
Anatomy
The average pancreas weighs between 75 and 125 g and measures 10 to 20 cm. It lies in the retroperitoneum just anterior to the first lumbar vertebrae and is anatomically divided into four portions, the head, neck, body and tail. The head lies to the right of midline within the C loop of the duodenum, immediately anterior to the vena cava at the confluence of the renal veins. The uncinate process extends from the head of the pancreas behind the superior mesenteric vein (SMV) and terminates adjacent to the superior mesenteric artery (SMA). The neck is the short segment of pancreas that immediately overlies the SMV. The body and tail of the pancreas then extend across the midline, anterior to Gerota’s fascia and slightly cephalad, terminating within the splenic hilum (Fig. 56-1).
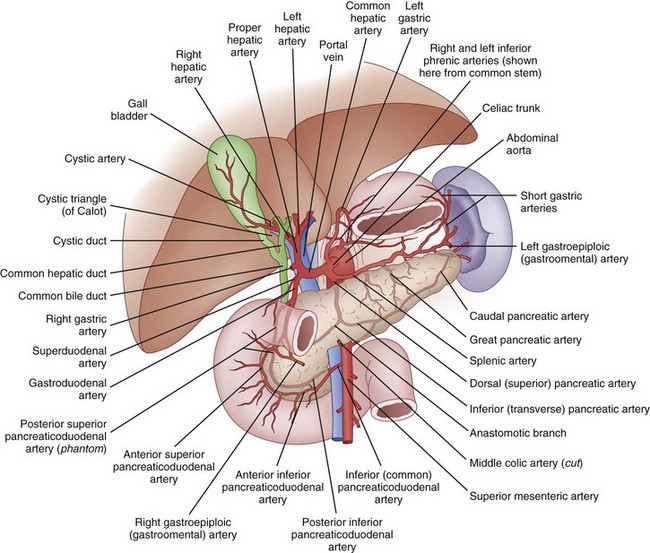
FIGURE 56-1 Anatomy.
(Netter illustration from www.netterimages.com. © Elsevier Inc. All rights reserved.)
Embryology
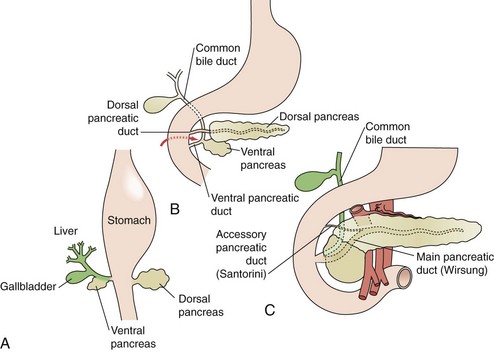
The exocrine pancreas begins development during the fourth week of gestation. Pluripotent pancreatic epithelial stem cells give rise to exocrine and endocrine cell lines, as well as the intricate pancreatic ductal network. Initially, dorsal and ventral buds appear from the primitive duodenal endoderm (Fig. 56-2A). The dorsal bud typically appears first and ultimately develops into the superior head, neck, body, and tail of the mature pancreas. The ventral bud develops as part of the hepatic diverticulum and maintains communication with the biliary tree throughout development. The ventral bud will become the inferior part of the head and uncinate process of the gland. Between the fourth and eighth week, the ventral bud rotates posteriorly in a clockwise fashion to fuse with the dorsal bud (see Fig. 56-2B). At approximately 8 weeks’ gestation, the dorsal and ventral buds are fused (see Fig. 56-2C).
The initiation of pancreas bud formation and differentiation of the ventral bud from the hepatic-biliary fates is dependent on the expression of pancreatic duodenal homeobox 1 (PDX1) protein and pancreas-specific transcription factor 1 (PTF1). In the absence of PDX1 expression in mice, pancreatic agenesis occurs, indicating its importance in the early phases of organogenesis. PTF1 expression is first detectable shortly after PDX1 in cells of the early endoderm, which will become the dorsal and ventral pancreas. By lineage analysis, 95% of acinar cells express PTF1. In PTF1 null mice, acini do not form. The notch signaling pathway is also critical to duct and acinar differentiation. In the absence of notch signaling, embryonic cells commit to endocrine lineage, suggesting that notch signaling is vital to exocrine differentiation. In addition to PDX1, PTF1, and notch signaling, complex interactions between mesenchymal growth factors such as transforming growth factor-β (TGF-β) and other signaling pathways, including hedgehog and Wnt, seem to play critical roles in pancreas development.1 The precise interactions that lead to normal organogenesis continue to be defined. Table 56-1 summarizes the factors and pathways that affect pancreas development.
Table 56-1 Molecular Factors and Pathways Associated With Pancreatic Organogenesis
| MUTATION | RELEVANCE |
|---|---|
| PDX1 | Critical role in exocrine differentiation; knockout mice develop primitive pancreatic buds, but agenesis of the organ. |
| PTF1 | Coexpression with PDX1 determines progenitor cells to pancreatic fate. |
| Notch signaling pathway | Suppresses endocrine differentiation, promoting exocrine development. |
| Hedgehog signaling pathway | Inhibition of hedgehog in PDX1-positive cells leads to initiation of endoderm differentiation into pancreas lineage. |
| Wnt signaling pathway | Complex Wnt signaling is important in all aspects of pancreas development; lack of Wnt signaling results in absence of acinar tissue. |
Pancreas Divisum
During normal organogenesis, the dorsal and ventral buds most commonly fuse to form a common duct, which enters the duodenum along with the common bile duct via the ampulla of Vater. Failure of the dorsal and ventral ducts to fuse during embryogenesis leads to pancreas divisum, a condition identified by a ventral pancreatic duct and common bile duct, which enter the duodenum via a major papilla, whereas a dorsal pancreatic duct enters through a minor papilla, which is slightly proximal (Fig. 56-3). Because most pancreatic exocrine secretions exit via the dorsal duct, pancreas divisum can lead to a condition of partial obstruction caused by a small minor papilla, leading to chronic backpressure in the duct. This relative outflow obstruction has been implicated in the development of relapsing acute or chronic pancreatitis. Although 10% of the population is affected by pancreas divisum, only rarely do affected individuals develop pancreatitis.
Physiology
Major Components of Pancreatic Juice
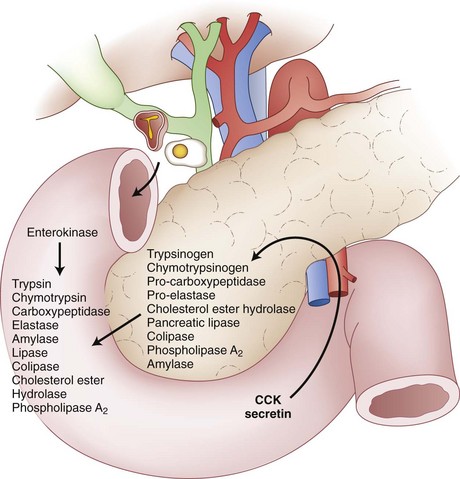
The main function of the exocrine pancreas is to provide the vast majority of the enzymes needed for alimentary digestion. Acinar cells synthesize many enzymes (proteases) that digest food proteins such as trypsin, chymotrypsin, carboxypeptidase, and elastase. Under physiologic conditions, acinar cells synthesize these proteases as inactive proenzymes that are stored as intracellular zymogen granules. With stimulation of the pancreas, these proenzymes are secreted into the pancreatic duct and eventually the duodenal lumen. The duodenal mucosa synthesizes and secretes enterokinase, which is the critical enzyme in the enzymatic activation of trypsin from trypsinogen.2 Trypsin also plays an important role in protein digestion by propagating pancreatic enzyme activation through autoactivation of trypsinogen and other proenzymes, such as chymotrypsinogen, procarboxypeptidase, and proelastase. Figure 56-4 summarizes the mechanisms of pancreatic exocrine secretion.
In addition to HCO3−, CO2 hydration also generates H+ ions, which are secreted by Na+ and H+ exchangers present in the basolateral membrane of ductal cells. These exchangers belong to the SLC9 gene family. The main function of these exchangers is to maintain the intracellular pH within a physiologic range. In addition, the basolateral membrane of duct cells contains multiple Na+,K+-ATPases that provide the primary force that drives HCO3− secretion; the Na+,K+-ATPase maintains the Na+ gradient used to extrude H+ as well. Finally, K+ channels present in the basolateral membrane of acinar cells maintain the membrane potential to allow recirculation of K+ ions brought by the Na+,K+ pump inside the cell. Figure 56-5 illustrates HCO3− secretion inside pancreatic duct cells.
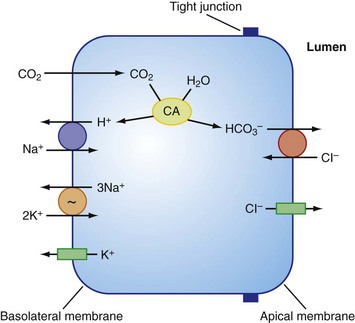
FIGURE 56-5 Cellular mechanism proposed for HCO3− secretion by pancreatic duct epithelium.
(From Steward MC, Ishiguro H, Case RM: Mechanisms of bicarbonate secretion in the pancreatic duct. Annu Rev Physiol 67:377–409, 2005.)
Acute Pancreatitis
The incidence of acute pancreatitis (AP) has increased during the past 20 years. AP is responsible for more than 300,000 hospital admissions annually in the United States. Most patients develop a mild and self-limited course; however, 10% to 20% of patients have a rapidly progressive inflammatory response associated with prolonged length of hospital stay and significant morbidity and mortality. Patients with mild pancreatitis have a mortality rate of less than 1% but, in severe pancreatitis, this increases up to 10% to 30%.3 The most common cause of death in this group of patients is multiorgan dysfunction syndrome. Mortality in pancreatitis has a bimodal distribution; in the first 2 weeks, also known as the early phase, the multiorgan dysfunction syndrome is the final result of an intense inflammatory cascade triggered initially by pancreatic inflammation. Mortality after 2 weeks, also known as the late period, is often caused by septic complications.4
Pathophysiology
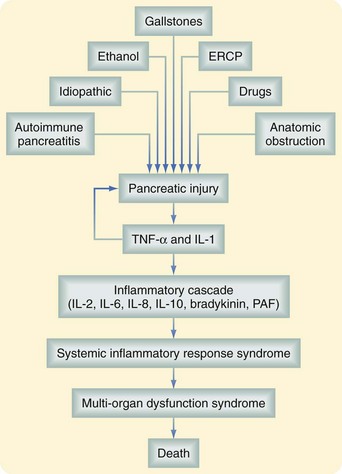
The exact mechanism whereby predisposing factors such as ethanol and gallstones produce pancreatitis is not completely known. Most researchers believe that AP is the final result of abnormal pancreatic enzyme activation inside acinar cells. Immunolocalization studies have shown that after 15 minutes of pancreatic injury, both zymogen granules and lysosomes colocalize inside the acinar cells. The fact that zymogen and lysosome colocalization occurs before amylase level elevation, pancreatic edema, and other markers of pancreatitis are evident suggests that colocalization is an early step in the pathophysiology and not a consequence of pancreatitis. In addition, the inflammatory response seen in AP can be prevented if acinar cells are pretreated with cathepsin B inhibitors. In vivo studies have also shown that cathepsin B knockout mice have a significant decrease in the severity of pancreatitis.2
Intra-acinar pancreatic enzyme activation induces autodigestion of normal pancreatic parenchyma. In response to this initial insult, acinar cells release proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukins (IL)-1, -2, and -6, and anti-inflammatory mediators such as IL-10 and IL-1 receptor antagonist. These mediators do not initiate pancreatic injury but propagate the response locally and systemically. As a result, TNF-α, IL-1, and IL-7, neutrophils, and macrophages are recruited into the pancreatic parenchyma and cause the release of more TNF- α, IL-1, IL-6, reactive oxygen metabolites, prostaglandins, platelet-activating factor, and leukotrienes. The local inflammatory response further aggravates the pancreatitis because it increases the permeability and damages the microcirculation of the pancreas. In severe cases, the inflammatory response causes local hemorrhage and pancreatic necrosis. In addition, some of the inflammatory mediators released by neutrophils aggravate the pancreatic injury because they cause pancreatic enzyme activation.5
The inflammatory cascade is self-limited in approximately 80% to 90% of patients. However, in the remaining patients, a vicious cycle of recurring pancreatic injury and local and systemic inflammatory reaction persists. In a small number of patients, there is a massive release of inflammatory mediators to the systemic circulation. Active neutrophils mediate acute lung injury and induce the adult distress respiratory syndrome frequently seen in patients with severe pancreatitis. The mortality seen in the early phase of pancreatitis is the result of this persistent inflammatory response. A summary of the inflammatory cascade seen in AP is shown in Figure 56-6.
Risk Factors
Biliary or Gallstone Pancreatitis
Gallstone pancreatitis is the most common cause of AP in the West. It accounts for 40% of U.S. cases. The overall incidence of AP in patients with symptomatic gallstone disease is 3% to 8%. It is seen more frequently in women between 50 and 70 years of age. The exact mechanism that triggers pancreatic injury has not been completely understood, but two theories have been proposed.6 In the obstructive theory, pancreatic injury is the result of excessive pressure inside the pancreatic duct. This increased intraductal pressure is the result of continuous secretion of pancreatic juice in the presence of pancreatic duct obstruction. The second, or reflux, theory proposes that stones become impacted in the ampulla of Vater and form a common channel that allows bile salt reflux into the pancreas. Animal models have shown that bile salts cause direct acinar cell necrosis because they increase the concentration of calcium in the cytoplasm; however, this has never been proven in humans.2
Alcohol-Induced Injury
Excessive ethanol consumption is the second most common cause of AP worldwide. It accounts for 35% of cases and is more prevalent in young men (30 to 45 years of age) than in women. However, only 5% to 10% of patients who drink alcohol develop AP. Factors that contribute to ethanol-induces pancreatitis include heavy ethanol abuse (>100 g/day for at least 5 years), smoking, and genetic predisposition. As compared with nonsmokers, the relative risk of alcohol-induced pancreatitis in smokers is 4.9.7
Alcohol has a number of deleterious effects in the pancreas. It triggers proinflammatory pathways such as nuclear factor κB (NF-κB), which increase the production of TNF-α and IL-1. It also increases the expression and activity of caspases. Caspases are proteases that mediate apoptosis. In addition, alcohol decreases pancreatic perfusion, induces sphincter of Oddi spasm, and obstructs pancreatic ducts through the precipitation of proteins inside the ducts.8
Endoscopic Retrograde Cholangiopancreatography–Induced Pancreatitis
AP is the most common complication after ERCP, occurring in up to 5% of patients. AP occurs more frequently in patients who have undergone therapeutic procedures as compared with diagnostic procedures. It is also more common in patients who have had multiple attempts of cannulation, sphincter of Oddi dysfunction, and abnormal visualization of the secondary pancreatic ducts after contrast injection. The clinical course is mild in 90% to 95% of patients.8
Metabolic Factors
Hypercalcemia is postulated to induce pancreatic injury through the activation of trypsinogen to trypsin and intraductal precipitation of calcium, leading to ductal obstruction and subsequent attacks of pancreatitis. Approximately 1.5% to 13% of patients with primary hyperparathyroidism develop AP.8
Diagnosis
Patients with AP are typically hyperglycemic; they can also have leukocytosis and abnormal elevation of liver enzyme levels. The elevation of alanine aminotransferase levels in the serum in the context of AP confirmed by high pancreatic enzyme levels has a positive predictive value of 95% in the diagnosis of acute biliary pancreatitis.6
Imaging Studies
Contrast-enhanced computed tomography (CT) is currently the best modality to evaluate the pancreas, especially if the study is performed using a multidetector CT scanner. The most valuable contrast phase to evaluate the pancreatic parenchyma is the portal venous phase (65 to 70 seconds after contrast injection), which allows evaluation of the viability of the pancreatic parenchyma amount of peripancreatic inflammation and presence of intra-abdominal free air or fluid collections. Noncontrast CT scanning may also be of value in the setting of renal failure by identifying fluid collections and/or extraluminal air.9
Abdominal magnetic resonance imaging (MRI) is also useful to evaluate the extent of necrosis, inflammation, and presence of free fluid. However, its cost and availability, and the fact that patients requiring imaging are critically ill and need to be in intensive care units, limit its applicability in the acute phase. Although magnetic resonance cholangiopancreatography (MRCP) is not indicated in the acute setting of AP, it has an important role in the evaluation of patients with unexplained or recurrent pancreatitis because it allows complete visualization of the biliary and pancreatic duct anatomy. In addition, IV administration of secretin increases pancreatic duct secretion, which causes a transient distention of the pancreatic duct. For example, secretin MRCP is useful in patients with AP and no evidence of a predisposing condition to rule out pancreas divisum, intraductal papillary mucinous neoplasm (IPMN), or the presence of a small tumor in the pancreatic duct.9
Assessment of Severity of Disease
The earliest scoring system designed to evaluate the severity of AP was introduced by Ranson and colleagues in 1974.10 It predicts the severity of the disease based on 11 parameters obtained at the time of admission and/or 48 hours later. The mortality rate of AP directly correlates with the number of parameters that are positive. Severe pancreatitis is diagnosed if three or more of the Ranson criteria are fulfilled. The main disadvantage is that it does not predict the severity of disease at the time of the admission because six parameters are only assessed after 48 hours of admission. Ranson’s score has a low positive predictive value (50%) and high negative predictive value (90%). Therefore, it is mainly used to rule out severe pancreatitis or predict the risk of mortality.11 The original scoring symptom designed to predict the severity of the disease and its modification for acute biliary pancreatitis are shown in Boxes 56-1 and 56-2.
Box 56-1 Ranson’s Prognostic Criteria for Nongallstone Pancreatitis
Ranson score ≥3 defines severe pancreatitis.
Box 56-2 Ranson’s Prognostic Criteria for Gallstone Pancreatitis
Ranson score ≥3 defines severe pancreatitis.
AP severity can also be addressed using the Acute Physiology and Chronic Health Evaluation (APACHE II) score. Based on the patient’s age, previous health status, and 12 routine physiologic measurements, APACHE II provides a general measure of the severity of disease. An APACHE II score of 8 or higher defines severe pancreatitis. The main advantage is that it can be used on admission and repeated at any time. However, it is complex, not specific for AP, and based on the patient’s age, which easily upgrades the AP severity score. APACHE II has a positive predictive value of 43% and a negative predictive value of 89%.11
Using imaging characteristics, Balthazar and associates12 have established the CT severity index. This index correlates CT findings with the patient’s outcome. The CT severity index is shown in Table 56-2.
Table 56-2 Computed Tomography Severity Index (CTSI) for Acute Pancreatitis
| FEATURE | POINTS |
|---|---|
| Pancreatic Inflammation | |
| Normal pancreas | 0 |
| Focal or diffuse pancreatic enlargement | 1 |
| Intrinsic pancreatic alterations with peripancreatic fat inflammatory changes | 2 |
| Single fluid collection/or phlegmon | 3 |
| Two or more fluid collections or gas, in or adjacent to the pancreas | 4 |
| Pancreatic Necrosis | |
| None | 0 |
| ≤30% | 2 |
| 30%-50% | 4 |
| >50% | 6 |
CTSI, 0-3, mortality 3%, morbidity 8%; CTSI, 4-6, mortality 6%, morbidity 35%; CTSI, 7-10, mortality 17%, morbidity 92%.
In 1992, the International Symposium on Acute Pancreatitis defined severe pancreatitis as the presence of local pancreatic complications (necrosis, abscess, or pseudocyst) or any evidence of organ failure. Severe pancreatitis is diagnosed if there is any evidence of organ failure or a local pancreatic complication (Box 56-3).
Box 56-3 Atlanta’s Criteria for Acute Pancreatitis
Organ Failure As Defined
Shock (systolic blood pressure <90 mm Hg)
Pulmonary insufficiency (PaO2 <60 mm Hg)
Renal failure (creatinine level >2 mg/dL after fluid resuscitation)
Systemic Complications
Disseminated intravascular coagulation (platelet count ≤100,000)
Fibrin split products >80 µg/dL
Treatment
There is no proven benefit in treating AP with antiproteases (e.g., gabexate mesilate, aprotinin), platelet-activating factor inhibitors (e.g., lexipafant), or pancreatic secretion inhibitors.3
Given the significant increase in mortality associated with septic complications in severe pancreatitis, a number of physicians advocated the use of prophylactic antibiotics in the 1970s. Recent meta-analyses and systematic reviews that have evaluated multiple randomized control trials have proven that prophylactic antibiotics do not decrease the frequency of surgical intervention, infected necrosis, or mortality in patients with severe pancreatitis. In addition, they are associated with gram-positive cocci infection such as by Staphylococcus aureus, and Candida infection, which is seen in 5% to 15% of patients.13
Special Considerations
Laparoscopic Cholecystectomy
In the absence of definitive treatment, 30% of patients with acute biliary pancreatitis will have recurrent disease. With the exception of older patients and those with poor performance status, laparoscopic cholecystectomy is indicated for all patients with mild acute biliary pancreatitis.3 Studies have shown that early laparoscopic cholecystectomy, defined as laparoscopic cholecystectomy during the initial admission to the hospital, is a safe procedure that decreases recurrence of the disease.6 Choledocolithiasis can be excluded via intraoperative cholangiography, ERCP, or laparoscopic common bile duct exploration.
For patients with severe pancreatitis, early surgery may increase the morbidity and length of stay.14 Current recommendations suggest conservative treatment for at least 6 weeks before laparoscopic cholecystectomy is attempted in this setting. This approach has significantly decreased morbidity.6
Complications
Sterile and Infected Peripancreatic Fluid Collections
The presence of acute abdominal fluid during an episode of AP has been described in 30% to 57% of patients.3 In contrast to pseudocysts and cystic neoplasias of the pancreas, fluid collections are not surrounded or encased by epithelium or fibrotic capsule. Treatment is supportive because most fluid collections will be spontaneously reabsorbed by the peritoneum. The presence of fever, elevated white blood cell (WBC) count, and abdominal pain suggest infection of this fluid and percutaneous aspiration is confirmatory. Percutaneous drainage and IV administration of antibiotics should be instituted if infection is present.
Pancreatic Necrosis and Infected Necrosis
Pancreatic necrosis is the presence of nonviable pancreatic parenchyma or peripancreatic fat; it can present as a focal area or diffuse involvement of the gland. Contrast-enhanced CT is the most reliable technique to diagnose pancreatic necrosis. It is typically seen as areas of low attenuation (<40 to 50 HU) after the injection of IV contrast. Normal parenchyma usually has a density of 100 to 150 HU.9 Up to 20% of patients with AP develop pancreatic necrosis. It is important to identify and provide proper treatment of this complication because most patients who develop multiorgan failure have necrotizing pancreatitis; pancreatic necrosis has been documented in up to 80% of the autopsies of patients who died after an episode of AP.4
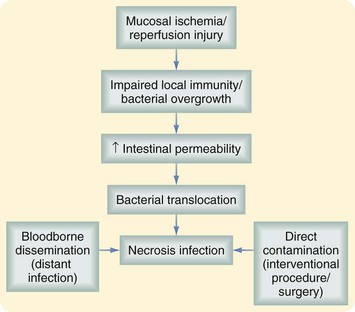
The main complication of pancreatic necrosis is infection. The risk is directly related to the amount of necrosis; in patients with pancreatic necrosis involving less than 30% of the gland, the risk of infection is 22%. The risk is 37% for patients with pancreatic necrosis that involves 30% to 50% of the gland and up to 46% if more than 70% of the gland is affected.4 This complication is associated with bacterial translocation usually involving enteric flora, such as gram-negative rods (e.g., Escherichia coli, Klebsiella and Pseudomonas spp.) and Enterococcus spp.
Infected pancreatic necrosis should be suspected in patients with prolonged fever, elevated WBC count, or progressive clinical deterioration. Evidence of air within the pancreatic necrosis seen on a CT scan confirms the diagnosis but is a rare finding. If infected necrosis is suspected, fine-needle aspiration (FNA) should be performed. A positive Gram stain and/or culture establish the diagnosis. Although positive cultures are confirmatory, a recent review has demonstrated that despite negative preoperative cultures, 42% of patients with so-called persistent unwellness will have infected necrosis.15 Figure 56-7 illustrates the pathophysiology of pancreatic necrosis infection.
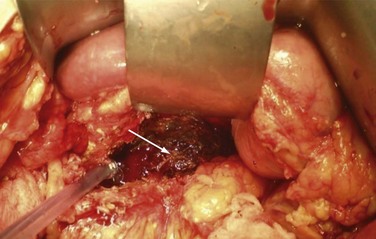
Definitive treatment for infected pancreatic necrosis is surgical débridement with necrosectomy, closed continuous irrigation, and open packaging (Fig. 56-8). The overall mortality rate after open necrosectomy is 25% to 30%.15 Outcomes are time-dependent, with patients who undergo surgery in the first 14 days having a mortality rate of 75%; those who undergo surgery between 15 and 29 days and after 30 days have mortality rates of 45% and 8%, respectively.16 As a result of the elevated morbidity and mortality rates with open débridement, endoscopic and laparoscopic techniques are being used more often. Both may ultimately provide similar outcomes, with hopes of reducing perioperative morbidity and mortality, although level 1 data are lacking.
Pancreatic Pseudocysts
Pancreatic pseudocysts occur in 5% to 15% of patients who have peripancreatic fluid collections after AP. By definition, the capsule of a pseudocyst is composed of collagen and granulation tissue and it is not lined by epithelium.17 The fibrotic reaction typically requires at least 4 to 8 weeks to develop. Figure 56-9 shows CT scans of a large pseudocyst arising in the tail of the pancreas.
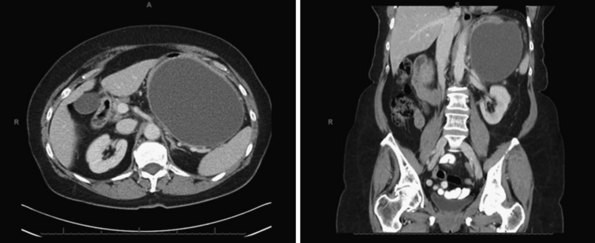
Observation is indicated for asymptomatic patients because spontaneous regression has been documented in up to 70% of cases; this is particularly true for patients with pseudocysts smaller than 4 cm in diameter, located in the tail, and no evidence of pancreatic duct obstruction or communication with the main pancreatic duct.17 Invasive therapies are indicated for symptomatic patients or when the differentiation between a cystic neoplasm and pseudocyst is not possible. Because most patients are treated with decompressive procedures and not with resection, it is imperative to have a pathologic diagnosis. Surgical drainage has been the traditional approach for pancreatic pseudocysts. However, there is increasing evidence that transgastric and transduodenal endoscopic drainage are safe and effective approaches for patients with pancreatic pseudocysts in close contact (defined as <1 cm) with the stomach and duodenum, respectively. In addition, transpapillary drainage can be attempted in pancreatic pseudocysts communicating with the main pancreatic duct. For patients in whom a pancreatic duct stricture is associated with a pancreatic pseudocyst, endoscopic dilation and stent placement are indicated.
Surgical drainage is indicated for patients with pancreatic pseudocysts that cannot be treated with endoscopic techniques and patients who fail endoscopic treatment. Definitive treatment depends on the location of the cyst. Pancreatic pseudocysts closely attached to the stomach should be treated with a cystgastrostomy. In this procedure, an anterior gastrostomy is performed. Once the pseudocyst is located, it is drained through the posterior wall of the stomach using a linear stapler. The defect in the anterior wall of the stomach is closed in two layers. Pancreatic pseudocysts located in the head of the pancreas that are in close contact with the duodenum are treated with a cystoduodenostomy. Finally, some pseudocysts are not in contact with the stomach or duodenum. The surgical treatment for these patients is a Roux-en-Y cystojejunostomy. Surgical cyst enterostomy is successful in achieving immediate cyst drainage in over 90% of cases. Following initial resolution, recurrent pseudocyst formation may occur in up to 12% of cases during long-term follow-up, depending on the location of the cyst and underlying cause of the disease.17
Pancreatic Ascites and Pancreaticopleural Fistulas
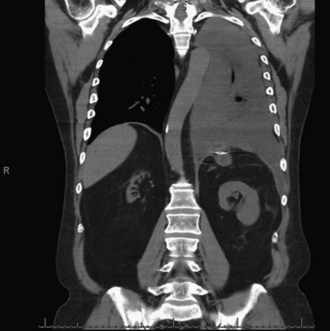
Posterior pancreatic duct disruption into the pleural space has been described rarely. Symptoms that suggest this condition include dyspnea, abdominal pain, cough, and chest pain. The diagnosis is confirmed with chest x-ray, thoracentesis, and CT scan. Figure 56-10 demonstrates a large, left-sided pleural effusion caused by a pancreatic-pleural fistula. Amylase levels above 50,000 IU in the pleural fluid confirm the diagnosis. It is more common after alcoholic pancreatitis and, in 70% of patients, is associated with pancreatic pseudocysts. Initial treatment requires chest drainage, parenteral nutritional support, and administration of octreotide. Up to 60% of patients respond to this therapy. Persistent drainage should also be treated with endoscopic sphincterotomy and stent placement. Patients who do not respond to these measures require surgical treatment, similar to that described for pancreatic ascites.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


