Epigenetic Changes in Lung Cancer: Pathobiological and Clinical Aspects
Epigenetic Changes in Lung Cancer: Pathobiological and Clinical Aspects
Ite A. Laird-Offringa
Within a decade after the publication of the first human genome sequence,1 and even before a full understanding of all its implications has been attained, a new frontier has emerged: epigenetics.2 The study of factors superimposed on the genes, or “epi”genetics, focuses on mitotically heritable modifications of DNA and histones, and the associated chromatin components that affect gene expression without altering gene sequence. 3 Epigenetics is one of the most exciting new frontiers in genome analysis. Two of the most widely studied epigenetic modifications are DNA methylation and histone modification. Many of the interacting proteins that bind directly or indirectly to methylated DNA or modified histones catalyze the formation or removal of other alterations, forming a complex regulatory network that is only beginning to be deciphered. 3,4,5,6 It has become clear that epigenetic deregulation contributes very importantly to cancer development and progression. 7,8,9,10,11 Profound epigenetic changes are seen in all cancer types, including lung cancer. 12,13,14,15,16 Epigenetic alterations in lung cancer show potential as molecular markers that could be applied to early detection, tumor classification, risk assessment, prognostication, and monitoring of cancer recurrence. 17,18,19,20 In addition, understanding the consequences of epigenetic changes can help dissect the molecular basis of lung cancer, providing new focal points for targeted therapies.
One of the most exciting aspects of epigenetic changes is their inherent reversibility. This has encouraged the development of novel drugs for cancer treatment, such as histone deacetylase inhibitors (HDACI) and DNA methylation inhibitors. 21,22 A number of these drugs are in clinical trials for numerous cancers, including those of the lung. With the advent of ever more powerful tools for genome-wide assessment, 23 our understanding of the lung cancer epigenome and its application to diagnosis and treatment promises to increase dramatically in the years to come. 7 Here, the basic concepts of epigenetics will be reviewed, and our current knowledge concerning epigenetic alterations in lung cancer will be discussed, including the type of changes identified and their pathological and clinical implications. Given the very large number of epigenetic alterations analyzed to date and the dramatic acceleration in acquired data, it is impossible to be comprehensive in one short chapter. Therefore, the important advances made in lung cancer research will be illustrated based on a limited number of key examples, and reviews will be cited throughout as a source of more detailed information.
GENETIC AND EPIGENETIC INTERACTIONS
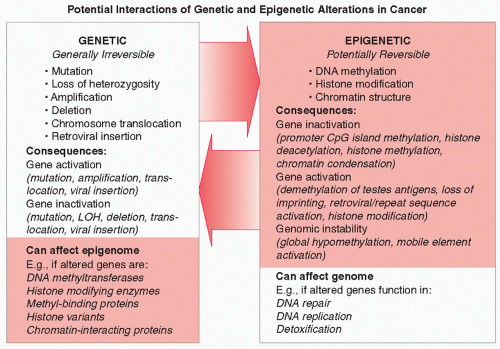
Initial research into the molecular basis of lung cancer focused on genetic alterations, such as mutations, loss of heterozygosity, deletions, and gene amplification. 24,25 Examples of genetic alterations in lung cancer include mutations in KRAS and the epidermal growth factor receptor (EGFR), loss of heterozygosity at chromosome 3p, and MYC gene amplification. However, it has become abundantly clear that epigenetic alterations contribute equally importantly to lung cancer development and progression. 12,13,14 Epigenetic alterations seen in lung cancer consist of DNA methylation changes (both loss and gain of methylation), changes in histone modifications, and alterations in chromatin structure and chromatin-associated proteins. Interactions between genetic and epigenetic hits in cancer cells can result in further alterations, 2,11,26,27 as outlined in Figure 7.1. For example, genetic alterations in the genes encoding components of the epigenetic machinery (such as DNA methyltransferases and HDACs) can affect the activity of these enzymes and thereby the transcriptional activity of many additional genes. In numerous cancers, including lung cancer, somatic changes in parts of the epigenetic machinery are seen.27 This potential for genetic alterations to affect epigenetics is underscored by the reported link between lung cancer risk and genetic polymorphisms in several genes encoding epigenetic enzymes. 27 Conversely, epigenetic alterations can lead to further genetic damage. For example, hypermethylation of DNA repair genes or genes encoding detoxification enzymes can affect the cell’s susceptibility to mutagenesis and could result in the genetic (in)activation of additional genes. 26 DNA methylation of 6-O-methylguanine DNA methyltransferase (MGMT), an enzyme involved in repair of alkylated guanine, is commonly seen in lung cancer. 15 Inactivation of MGMT has been linked to an increase in RAS gene mutation frequency. 28 In support of their potential to affect cancer development, polymorphisms in MGMT and other DNA repair genes have been linked to lung cancer risk in various populations. 29,30,31 These examples illustrate that genetic and epigenetic changes should not be seen as independent but as components of a complex interactive network that is responsible for the development and progression of lung cancer (Fig. 7.1). Combined analysis of both types of molecular changes will accelerate the elucidation of the molecular pathways affected in lung cancer, and could be especially helpful in characterizing particular types of lung cancer (e.g., histological subtypes or lung cancer from smokers vs. nonsmokers). This holistic view of (epi)genetic alterations is also highly relevant to the clinic, as the use of certain cytotoxic drugs may potentiate or inhibit the efficacy of epigenetic drugs and vice versa. 21,22
FIGURE 7.1 Interaction between genetic and epigenetic alterations in cancer. Left panel: Genetic “hits,” which are generally irreversible and can result in activation or inactivation of the altered gene. If such a gene encodes a product involved in epigenetic regulation, like a histone methyltransferase, a DNA methyl-binding protein, 67 a histone isoform, an enzyme that adds or removes histone modifications, or a protein that interacts with such modifications (transcriptional regulators, coactivators, or corepressors), this could result in epigenetic alterations. Right panel: Epigenetic hits are potentially reversible, and when they occur in genes that affect the integrity of the genome, such as DNA repair genes or genes encoding proteins involved in DNA replication or detoxification, they can increase the likelihood of acquisition of additional genetic alterations.
DNA METHYLATION
In mammals, DNA methylation occurs at the 5-position of cytosine, in the context of a mini palindrome: a cytosine-phosphateguanine (CpG) dinucleotide. The palindromic nature of methylation allows the propagation of this modification following DNA replication. In the normal mammalian genome, some areas are heavily methylated, such as sections of the inactive X chromosome in women, pericentromeric regions, and parentally imprinted genes. Indeed, DNA methylation is essential for proper development and viability. 3 Methylation in mammals is carried out by at least three enzymes, the maintenance DNA methyltransferase DNMT1 (which methylates daughter strands following DNA replication) and de novo DNA methyltransferases DNMT3A and 3B. 32 All three genes are essential, as illustrated by mouse knockout experiments. 32 A large number of splice isoforms exists, a number of which appear to target particular genes or areas of the genome and some of which are implicated in cancer. 33,34 In lung cancer, overexpression of the deltaDNMT3B4 variant correlates strongly with RASSF1A methylation, and knockdown of this methyltransferase resulted in a rapid demethylation of the RASSF1A CpG island. 35 This effect was gene-specific, as no changes in methylation of CDKN2A were observed.
CpG dinucleotides exist in two general environments in normal cells: sparsely distributed and clustered. On the one hand, CpGs are sprinkled throughout the genome, and these CpGs are usually methylated. Spontaneous deamination of methyl-C results in thymine, which is less efficiently repaired than the uracil resulting from deamination of unmethylated cytosine. This has resulted in depletion over time of CpGs in areas that are usually methylated. 36 Thus, the remaining dense clusters of CpGs, called CpG islands, 37 are presumed to be normally unmethylated. It is estimated that 40% of human genes contain such CpG islands in their promoter regions. 1
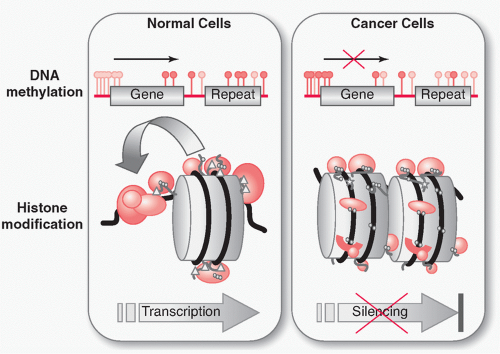
In cancer, a profound disruption of DNA methylation is seen (see Fig. 7.2, top). 7,8,9,10,11 Global hypomethylation occurs, which has been proposed to occur very early during cancer development and results in a net loss of methyl-C. This is thought to contribute to carcinogenesis in two possible ways: the transcriptional activation of previously methylated sequences and the loss of chromosome stability. In contrast, the local hypermethylation at promoter CpG islands contributes to carcinogenesis through gene inactivation, silencing a wide variety of growth control and tumor suppressor genes, such as genes involved in growth, adhesion, apoptosis, cell cycle, differentiation, signaling, and transcription.
FIGURE 7.2 Epigenetic abnormalities in cancer. In nontumor cells (left), CpG islands are generally unmethylated (gray lollipops), while sporadic CpGs are usually methylated (black lollipops). In actively transcribed genes, the structure of chromatin is loose, allowing access of the transcriptional machinery to the promoter region. Acetylation of lysines (triangles) in the N-terminal tails of histones 3 and 4 reduces positive charge and relaxes the attraction to negatively charged DNA. Acetylation, monomethylation, dimethylation, and trimethylation (balls) and other modifications such as phosphorylation and sumoylation (stars) of the histone tails can mediate interactions directly or indirectly with the transcriptional machinery and with enzymes that can add further posttranslational modifications. In cancer cells (right), a genome-wide loss of DNA methylation at sporadic CpGs and previously methylated sequences such as repeats is seen. (In certain cases this can lead to gene activation, not shown.) Simultaneously, many promoter CpG islands become hypermethylated. This can result in silencing of tumor suppressor genes. Methyl-binding proteins interacting with methylated cytosines can recruit histone deacetylases, which can in turn lead to reduced chromatin access and transcriptional silencing. This model is a simplification; methylation, histone modification, and transcription are not always concordant—not all methylated genes are silenced, nor are all silent genes methylated.
DNA methylation is by far the best-studied epigenetic change in many cancers, including lung cancer. This is, on the one hand, because promoter CpG island hypermethylation is linked to gene silencing, and such silencing is thought to play a key role in the development and progression of cancer. On the other hand, DNA methylation has been extensively analyzed because it promises to provide powerful molecular markers for lung cancer. 14,15,38 Importantly, straightforward techniques exist to assess this modification. 39 Initially, analysis strategies were based on a target gene approach, utilizing DNA methylation-sensitive restriction enzymes and polymerase chain reaction (PCR)-based methods that rely on bisulfite conversion. Bisulfite conversion, a chemical treatment that converts unmethylated cytosine to uracil while methylated cytosine is protected,40 allows methylation information to be incorporated into the DNA sequence (otherwise it would be lost during PCR). Bisulfite-converted DNA can be analyzed by many methods, depending on the design and location of the PCR primers. The most common methods are bisulfite genomic sequencing, methylation-specific PCR (MSP), and its real-time version, MethyLight or its variation quantitative MSP (QMSP). Bisulfite genomic sequencing consists of amplification followed by cloning and sequencing and provides information on the methylation status of all the Cs on the same DNA strand in the amplified area. 41 MSP utilizes primers that cover a number of methylation sites, 42 allowing interrogation of one or more CpGs in a small area. MethyLight incorporates the inclusion of a fluorescent probe between the primers, enabling real-time PCR detection, and includes control reactions on fully methylated DNA (treated with Sss I enzyme), allowing quantitative measurement of methylation. 43,44 More recently, higher throughput and more genome-wide approaches have been developed, such as restriction landmark genomic scanning 45 and specific amplification/purification of methylated versus unmethylated DNA (using restriction enzymes46 or binding enrichment)47,48 followed by probing of microarrays. Expression profiling of cancer cell lines treated with demethylating drugs has also been used to identify DNA methylation-silenced genes (e.g., in non-small cell lung cancer [NSCLC]).49 In the future, direct sequencing with high-throughput methods 23 using either methylation-enriched or bisulfite-treated DNA will be applied. Although reports based on these latter methods are beginning to be published,50 these techniques are in their infancy and many technical hurdles remain. In addition, they are extremely costly. At this time, high-throughput bead-based PCR methods combine the best of both worlds in richness of data and affordability, allowing the reproducible and rapid interrogation of thousands of targeted loci (Illumina Inc., GoldenGate platform,).51 This approach was successfully applied to lung adenocarcinoma (Table 7.1)51 and has recently been further developed to provide close to genome-wide representation of CpG islands (Illumina Inc., Infinium platform). All of these methods have yielded a great amount of information on DNA methylation changes in many cancers including lung cancer. This knowledge, and further epigenomic profiling, promise to change the way in which lung cancer is detected and treated.
Effects of Hypomethylation in Lung Cancer Because an overall hypomethylation is observed in cancer cells, it had originally been assumed that the cancer-causing effect of methylation changes was based on loss of promoter CpG island methylation resulting in proto-oncogene activation. 52 Indeed, loss of methylation can lead to gene activation in lung cancer, although the activated genes are not necessarily considered canonical proto-oncogenes. One category of such genes is the parentally imprinted genes—genes for which either the maternally or paternally inherited allele is normally methylated. Hypomethylation can result in loss of imprinting, thereby contributing to cancer development; biallelic expression of the normally imprinted insulin-like growth factor 2, mesodermspecific transcript and H19 genes has been seen in lung cancer and is thought to contribute to the carcinogenic phenotype. 53,54 Another type of gene that can be activated by hypomethylation is the family of testis-specific antigens—these genes are usually methylated and silent in all somatic tissues but the testes. 55 Expression of testis-specific antigens has been noted in many tumor types including lung cancer, and these antigens are seen as potential immunotherapy targets. 55,56,57 Loss of methylation of transposable elements and repeats is also observed in lung cancer 58 and can lead to mobility of such elements, causing further genetic damage. 11 In addition, read-through from such demethylated elements may result in the aberrant activation of neighboring genes. Hypomethylation might also play a role in the activation of microRNAs (miRNAs), many of which are deregulated in cancer. 59,60 In the lung, the normally methylated let-7a-3 miRNA was found to be hypomethylated in two out of eight lung adenocarcinomas and forced overexpression of this miRNA increased the oncogenic properties of lung cancer cell line A549. 61
Besides contributing to carcinogenesis through gene activation, a second consequence of hypomethylation is thought to be genomic instability. Mice genetically engineered to underexpress DNA methyltransferases show an increased frequency of loss of heterozygosity and an elevated incidence of hematopoietic malignancies. 62 Inactivation of DNMT1 and DNMT3b in a human colorectal cancer cell line led to aneuploidy. 63 However, there appears to be little evidence that hypomethylation is severely deleterious in this way in lung cancer. A recent analysis of methylation of five human squamous cell lung carcinomas and normal matched tissue showed prominent hypomethylation of repetitive elements but little methylation loss in single-copy sequences. 58 This supports the notion that the effect of hypomethylation in lung cancer might be limited and that clinical benefits might be achieved with methylationblocking therapies. Importantly, the leukemia-prone DNMT hypomorphic mice mentioned previously show a lower incidence of intestinal cancer, pointing to a protective effect of hypomethylation in certain tumor types. 64 Indeed, treatment of a mouse xenograft model for human lung cancer with DNA methylation and histone deacetylation inhibitors suppressed tumor growth without apparent toxicity. 65 A similar treatment of a murine lung cancer model cut lung tumor development in half, emphasizing the potential of epigenetic drugs for lung cancer treatment. 66
HYPERMETHYLATION IN LUNG CANCER: APPLICATION TO MARKER DEVELOPMENT
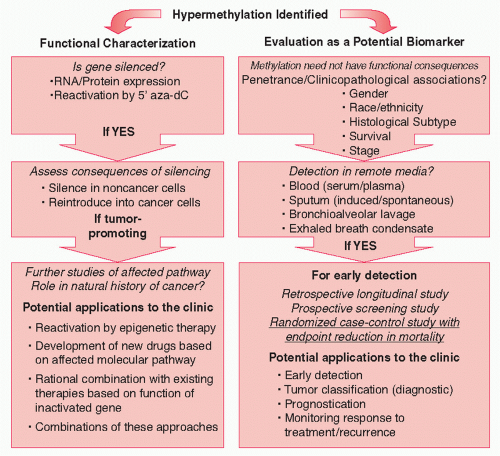
Although it would appear that the effects of hypomethylation in lung cancer are modest, hypermethylation of promoter CpG islands is widely observed. 12,13,14,15,16,38 Hypermethylation is associated with transcriptional shutdown. 3 This could happen directly through steric interference of methylated cytosines with transcription factor and cofactor binding sites, or indirectly, through the attraction of methyl-binding proteins to the DNA, which in turn recruit HDAC enzymes and other epigenetic modifiers (see Fig. 7.2). 67 In lung cancer, hundreds of studies have been devoted to the characterization of hypermethylation events. One of the driving forces behind this research is the desire to identify DNA methylation markers for early lung cancer detection. 14,15,38 DNA hypermethylation analyses could yield powerful candidate markers for lung cancer because only a small region of each gene needs to be interrogated, and DNA is a PCR-amplifiable substance that can be detected in bodily fluids. 14,15,38 Successful development of markers for cancer is a long process that should culminate in a randomized case-control study that demonstrates a reduction in mortality. 68 The process for the development of DNA methylation loci into markers for early lung cancer detection is diagrammed in the right panel of Figure 7.3.
TABLE 7.1 Epigenetic Profiling of DNA Methylation Loci in Lung Cancer
Screened 1536 CpG sites in 371 genes, identified 55-gene panel that is 92% sensitive and 100% specific on 12 AD and 12 matched AdjNTL
AD
Eight markers examined in detail by bisulfite genomic sequencing, all hypermethylated in 4/4 tumors vs. 2 normal lung samples ASCL2, CDH13, HOXA11, HOXA5, NPY, RUNX3, TERT, and TP73
bAD, adenocarcinoma; LuCa, mix of lung cancer types or no type specified; NSCLC, non-small cell lung cancer; SQ, squamous cell lung cancer.
cHuman Genome Organization name used unless none is available. Sensitivity and specificity numbers based on tumor tissues, not remote media.
Most DNA methylation studies in lung cancer have focused on NSCLC, which makes up about 85% of all lung cancers. Small cell lung cancer, a very aggressive cancer with poor survival, is considered by many to be a poor candidate for the development of early detection molecular makers due to the rapid progression of the disease. In contrast, NSCLC patients, which include the major groups adenocarcinoma (∽40%), squamous cell carcinoma (∽30%), large cell carcinoma (∽10%), and miscellaneous other histological subtypes such as carcinoids and neuroendocrine cancers (∽5%), 69 could benefit importantly if cancers that would normally lead to death could be detected at an early stage. 70 A comparison of methylation profiles of SCLC and NSCLC cell lines and tumors indicates that hypermethylation profiles are distinct for these two groups. 71,72,73 Not surprisingly, differences between hypermethylation profiles of NSCLC histological subtypes have also been observed, 15,20,72,74,75,76,77 meshing with other molecular and clinicopathological differences found in these tumor types. 78,79,80,81 This suggests that a panel of DNA methylation markers would be optimal, and that this panel should include pan-lung cancer markers as well as ones for distinct histological subtypes. Indeed, a panel of markers would be needed even for a single subtype because penetrance of molecular changes in cancer is usually less than 100%; it would be unexpected to find one marker with very high sensitivity and specificity. 20,74
FIGURE 7.3 Schematic outlining studies for functional characterization of gene hypermethylation (left) or for development of hypermethylated loci into lung cancer biomarkers (right).
The first step in molecular marker development is the identification of promising candidate markers. 68 In the case of DNA methylation markers for lung cancer, it is of high priority to identify frequently methylated genes or loci (we refer to the CpG island section we are probing as a locus, because a given gene can be probed in multiple areas within a single or multiple CpG islands). These loci should also show substantially increased methylation levels over those found in healthy tissues. Thus, the initial focus should be on penetrance and DNA methylation levels. Because even noncancerous lung tissue from long-term smokers may have accumulated substantial methylation caused by age and environmental exposure, 77,82,83,84,85 many labs, including ours, have chosen to compare cancer tissues to this type of “high-background” control tissue (referred to here as adjacent nontumor lung [AdjNTL]) (Table 7.1). This ensures that identified hypermethylation markers are indeed cancer-specific and not merely indicative of environmental exposure. (Comparison to healthy lung from nonsmokers would be of use for the development of risk markers or identification of candidates for chemoprevention treatments [perhaps even epigenetic ones], once these become available.)
Many of the genes studied early on did not show high methylation frequencies,15 but more recent efforts by several groups to examine much larger collections of genes have yielded a number of panels that might deliver high sensitivity and specificity, based on the examination of tissues (Table 7.1). 20,49,51,58,74,75,86,87,88,89,90,91,92 Some these panels contain genes that were identified early on (such as CDKN2A/p16, MGMT, and RASSF1), 74,75,88 but many new loci have been added to the repertoire, including homeotic genes involved in development, such as members of the HOX and PAX families.19,20,51,58,90,92 The latter group of methylated loci agrees with the observation that genes occupied in embryonic stem cells by transcriptionally repressive polycomb group complexes appear to be prone to methylation in cancer.93 The significance of methylation of these genes is unclear, since it is thought that they may already have been silent in noncancerous lung, but their involvement hints at the potential role of stem cells in lung cancer development.93 Whether the hypermethylation of potential DNA methylation markers is functional or not (i.e., leads to transcriptional silencing) is not relevant, as long as penetrance is high and hypermethylation is associated with the presence of cancer. Many of the marker panels in Table 7.1 must still be validated on independent tumor sets, and their ability to identify lung cancer independently of gender, histological subtype, racial/ethnic group, and/or stages of cancer must be further scrutinized (Fig. 7.3, right panel). Once that is accomplished, they can be taken to the next phase of marker development: clinical assay validation. 68 For these panels to function in early lung cancer detection, they must be detectable in patient remote media: bodily fluids that could carry methylated DNA molecules from the cancer and that could be sampled relatively noninvasively.
Only gold members can continue reading. Log In or Register to continue