Irwin Klein
Endocrine Disorders and Cardiovascular Disease
The endocrine system is tightly linked with many important cardiovascular diseases. As our understanding of the cellular and molecular effects of various hormones has evolved, we better understand the clinical manifestations that arise from excessive secretion of hormone and from glandular failure and subsequent hormone deficiency states. More than 200 years ago the English physician Caleb Hillier Parry described a woman with goiter and palpitations in whom “each systole shook the whole thorax.” He was the first to suggest a connection between diseases of the heart and enlargement of the thyroid gland. Recognition of the cardiovascular abnormalities associated with pathologic changes in endocrine glands preceded identification of the specific hormones produced by these glands. This chapter reviews the spectrum of cardiac disease states that arise from changes in specific endocrine function. This approach allows us to explore the cellular mechanisms whereby various hormones can alter the cardiovascular system through changes in lipid metabolism and actions on cardiac myocytes, vascular smooth muscle cells, and other target cells and tissues.
Pituitary Gland
The pituitary gland consists of two distinct anatomic portions. The anterior pituitary, or adenohypophysis, contains six different cell types; five of them produce polypeptide or glycoprotein hormones, and the sixth is composed of nonsecretory chromophobic cells. Of these cell types, the somatotropic cells, which secrete human growth hormone (hGH), and the corticotropic cells, which produce adrenocorticotropic hormone (ACTH), can contribute to cardiac disease. The posterior pituitary, or neurohypophysis, is the anatomic location of the nerve terminals that secrete vasopressin (antidiuretic hormone) to control water balance or oxytocin, the milk letdown polypeptide.
Growth Hormone
In adults, excessive secretion of hGH before fusion of the bony epiphysis leads to the clinical syndrome of gigantism, whereas increased secretion of hGH after maturation of the long bones leads to acromegaly. hGH exerts its cellular effects through two major pathways. The first is by binding of the hormone to specific hGH receptors on target cells. Such receptors have been identified in the heart, skeletal muscle, fat, liver, and kidneys, as well as in many additional cell types throughout fetal development.1 The second growth-promoting effect of hGH results from stimulation of the synthesis of insulin-like growth factor type I (IGF-I). This protein is synthesized primarily in the liver, but other cell types can produce IGF-I under the influence of hGH.
Shortly after identification of the IGF family, it was proposed that this second messenger mediates most actions of hGH. The clinical disease activity of patients with an excess of hGH (acromegaly) correlates better with serum levels of IGF-I than with hGH levels. The ability to promote glucose uptake and cellular protein synthesis gave rise to the term “insulin-like.” IGF-I binds to its cognate IGF-I receptor, which is located on almost all cell types. Genetic experiments have demonstrated that the presence of IGF-I receptors on cell types is closely linked to the ability of these cells to divide. Studies in which the IGF-I receptor was overexpressed in cardiac myocytes reportedly produced an increase in myocyte number and mitotic rate and enhanced the replication of postdifferentiated myocytes. Harnessing of this action could benefit genetic manipulation and, in conjunction with cardiac stem cells, could enhance repair of diseased myocardium.
Infusion of hGH or IGF-I acutely changes hemodynamics. The acute increases in cardiac contractility and cardiac output may result, at least in part, from a decrease in systemic vascular resistance and cardiac afterload.2 Short-term administration of hGH and IGF-I does not increase blood pressure.3,4
Cardiovascular Manifestations of Acromegaly
Acromegaly is a relatively uncommon condition (≈900 new cases each year in the United States). Acromegaly and pituitary-dependent human gigantism are associated with markedly increased morbidity and mortality, primarily from cardiovascular disease. Untreated acromegaly, identified by the characteristic clinical signs of acral growth and symptoms and by increased hGH secretion, markedly shortens life expectancy, with less than 20% of patients surviving beyond 60 years. Multiple studies have implicated increased neoplasia arising from the gastrointestinal tract, colon polyps, colon cancer, and pulmonary disease in this increased mortality,5 but cardiovascular and cerebrovascular effects, including hypertension, cardiomegaly, congestive heart failure, and cerebral vascular accidents, lead to the major events that limit survival.6,7
The cardiovascular and hemodynamic effects of acromegaly vary considerably depending on age and severity and duration of the disease.8 Patients in whom the condition was diagnosed with less than 5 years of disease activity had no significant abnormality in systolic or diastolic blood pressure, but echocardiographic evaluation showed that the left ventricular mass index increased almost 35% and the cardiac index increased 24%.9 Measures of systolic function, including stroke index, increased significantly, and systemic vascular resistance rose by 20% in patients with newly diagnosed, untreated acromegaly when compared with age- and sex-matched controls. Left ventricular diastolic function was normal.6 These studies contrast with reports that a longer duration of acromegaly produces left ventricular dysfunction and cardiomyopathy. In untreated acromegaly, global left ventricular diastolic dysfunction accompanies cardiac hypertrophy. The regional myocardial systolic strain abnormalities can be identified by Doppler imaging and can be reversed with treatment.10 A recent study identified impaired diastolic function in acromegaly, especially in older patients with diabetes mellitus.11
Known cardiac disease risk factors—including hypertension, insulin resistance, diabetes mellitus, and hyperlipidemia—frequently occur in patients with acromegaly. Although initial reports suggested that accelerated atherosclerosis causes impairment of cardiac function in patients with longstanding acromegaly, a postmortem study revealed significant coronary artery disease in only 11% of patients dying of disease-related causes. Angiography shows normal or dilated coronary arteries in most cases. Nuclear stress testing is positive in less than 25% of patients, thus indicating that atherosclerosis and ischemic heart disease are unlikely to account for the marked degree of biventricular cardiac hypertrophy, cardiac failure, and cardiovascular mortality. A rather specific functional and histologic change in myocytes appears to arise from the prolonged excessive serum levels of hGH and IGF-I.9 As many as two thirds of patients with acromegaly have echocardiographic criteria for left ventricular hypertrophy (LVH).6,8 Right ventricular mass also increases in acromegaly, a finding indicating a more generalized process beyond systemic hypertension.10 Asymmetric septal hypertrophy, initially thought to be common in patients with acromegaly, is an unusual finding. Acromegaly increases the prevalence of aortic and mitral valve disease, which persists despite cure of the acromegaly.12 Progressive mitral regurgitation and increased left ventricular preload and afterload occur in patients with uncontrolled acromegaly.13 Patients with acromegaly may exhibit dilation of the aortic root (aortic ectasia) and/or defects in the cardiac conduction system.8,12,14
Histologic evaluation of acromegalic cardiac tissue reveals an increase in myocyte size (hypertrophy) without an increase in cell number. Acromegaly is associated with interstitial fibrosis and infiltration of a variety of inflammatory cells, including mononuclear cells, consistent with myocarditis.6 Some of these histologic findings might result from IGF-I–promoted programmed cell death (apoptosis).
Functional changes accompany pathologic involvement of the heart in acromegaly.9,10 Although approximately 10% of patients with newly diagnosed acromegaly have signs and symptoms of cardiac compromise, this percentage increases markedly with longer disease duration.13–15 Some studies have reported a low (4%) incidence of overt left ventricular failure, which suggests that supervening factors, including hypertension, type 2 diabetes, and hyperlipidemia, are necessary to impair contractile function.16 In acromegaly, LVH and impaired left ventricular function can occur in those with longstanding disease but without hypertension, thus indicating that high levels of hGH and/or IGF-I can produce cardiac myopathic changes per se. Overt heart failure is associated with a poor prognosis.16 Therapy for acromegaly can prevent progression to heart failure.17,18
Abnormalities on the electrocardiogram (ECG), including left-axis deviation, septal Q waves, ST-T wave depression, abnormal QT dispersion, and conduction system defects, develop in up to 50% of patients with acromegaly. A variety of dysrhythmias can occur, including atrial and ventricular ectopic beats, sick sinus syndrome, and supraventricular and ventricular tachycardia.6 Fourfold increases in complex ventricular arrhythmias and late potentials are observed on signal-averaged ECGs; they are thought to be predictors of ventricular irritability and were also more common in active acromegaly than in treated patients.14 In contrast, exercise stress testing with electrocardiographic monitoring did not show inducible rhythm disturbances or evidence of ischemia, thus suggesting that the disturbances in left ventricular rhythm are not related to underlying ischemia. Cardiac autonomic function, as measured by heart rate recovery and variability, was altered in patients with newly diagnosed, untreated acromegaly.19
Secondary hypertension associated with acromegaly occurs in 20% to 40% of patients.5,6,8 Given the overall high prevalence rate of hypertension in the adult population and the insidious onset of acromegaly, determining whether hypertension is secondary or merely coincidental is difficult. Improvement in hypertension with therapy for acromegaly, however, suggests that they are related.17 Although observational studies of survival in patients with acromegaly initially suggested that hypertension was not an independent risk factor for mortality, a survey of patients dying of the disease found that their mean blood pressure was higher than that in those who survived.5 The mechanism underlying hypertension in acromegaly is not clearly understood. Patients with newly diagnosed acromegaly and short-duration disease had systolic and diastolic blood pressure no different from that in age- and sex-matched controls, but the cardiac index was significantly increased. In patients with longstanding acromegaly, arterial intimal thickness is increased, and these vascular changes respond to lowering of hGH levels.2
Administration of growth hormone promotes sodium retention and volume expansion and appears to have a potent antinatriuretic effect independent of any effect on aldosterone.3,18 Studies of the renin-angiotensin-aldosterone system have shown failure to inhibit release of renin optimally by volume expansion. Both angiotensin-converting enzyme inhibitors and angiotensin receptor blockers cause a paradoxical increase in blood pressure in patients with acromegaly. The role of hyperinsulinemia in the hypertension associated with acromegaly has been questioned. Increased serum insulin can contribute to urinary sodium retention, impairment of endothelial-dependent vasodilation, decreased nitric oxide production, and increased sympathetic activity.
Diagnosis
In 99% of cases, acromegaly arises from benign adenomas of the anterior pituitary gland.5,17 At diagnosis most of these neoplasms are classified as macroadenomas (>10 mm), and patients have historical clinical evidence of having had the disease for longer than 10 years. The diagnosis can be confirmed by demonstrating a serum hGH level higher than 5 ng/dL and a serum IGF-I level higher than 300 mIU/mL, measured 1 hour after a 100-g glucose load. In most patients, fasting hGH levels are higher than 10 ng/mL. Localization of the tumor can be established by magnetic resonance imaging (MRI) of the pituitary gland. Rarely, growth hormone–releasing hormone can be secreted and cause diffuse hyperplasia of the pituitary. Such changes must prompt consideration of a neoplastic lesion residing in other parts (ectopic) of the endocrine system.
Therapy
Transsphenoidal surgery with resection of the adenoma is the procedure of choice for initial management. If hGH and/or IGF-I levels remain elevated, radiotherapy in older patients or dopamine or somatostatin receptor agonists in younger patients can restore normal serum hGH and IGF-I levels. Octreotide acetate is a pharmacologic analogue of somatostatin that in the vast majority of patients, effectively lowers hGH to less than 5 ng/mL. Primary therapy might involve lowering IGF-I levels and shrinking tumor size in selected cases.17,20 The cardiovascular complications of acromegaly, including hypertension, LVH, and left ventricular dysfunction, improve with treatment, and survival is significantly better in patients achieving disease remission.7,8,13,20 Pegvisomant, a growth hormone receptor antagonist, can normalize IGF-I levels with long-term therapy and may play a role in somatostatin-resistant patients.14
Growth Hormone Deficiency
Childhood hGH deficiency is associated with increased body fat, central obesity, and an atherogenic lipid profile, as well as with impaired linear growth. Growth hormone replacement therapy appears to reverse or improve all these abnormalities.21
Adrenal Gland
Adrenocorticotropic Hormone and Cortisol
The adrenocorticotropic cells in the anterior pituitary synthesize a large protein (pro-opiomelanocortin), which is then processed within the corticotropic cell into a family of smaller proteins that include alpha-melanocyte–stimulating hormone, beta-endorphin, and ACTH. ACTH, in turn, binds to specific cells within the adrenal gland. Anatomically, the adrenal gland consists of two major segments, the cortex and the medulla. The cortex zona glomerulosa produces aldosterone, and the zona fasciculata produces primarily cortisol and some androgenic steroids. The zona reticularis also produces cortisol and androgens. ACTH regulates the synthesis of cortisol in the zona fasciculata and reticularis. The zona glomerulosa shows much less ACTH responsiveness and responds primarily to angiotensin II by increased secretion of aldosterone.
Cushing Disease
Excessive cortisol secretion and its attendant clinical disease states can arise from excessive release of ACTH by the pituitary (Cushing disease) or through the adenomatous or rarely malignant neoplastic process arising in the adrenal gland itself (Cushing syndrome). Well-characterized conditions of adrenal glucocorticoid and mineralocorticoid excess appear to result from the excessively high levels of (ectopic) ACTH produced by small cell carcinoma of the lung, carcinoid tumors, pancreatic islet cell tumors, medullary thyroid cancer, and other adenocarcinomas and hematologic malignancies.
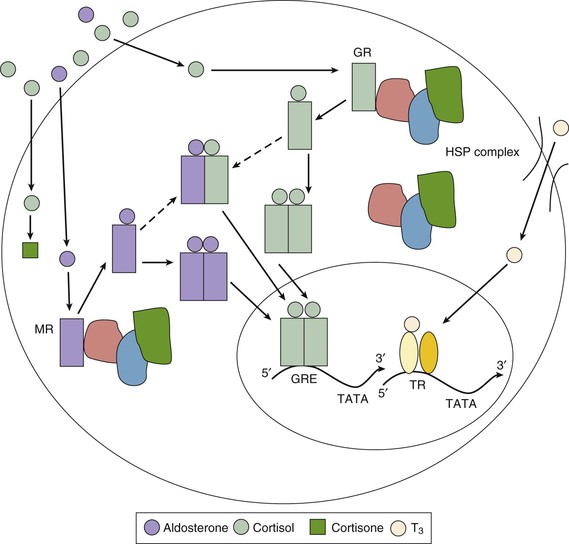
Cortisol, a member of the glucocorticoid family of steroid hormones, binds to monomeric receptors located within the cytoplasm of many cell types (Fig. 81-1). The unliganded glucocorticoid receptors are bound to heat shock protein complexes. After binding cortisol, the receptors dissociate from these complexes, homodimerize or occasionally heterodimerize, translocate to the nucleus, and function as transcription factors. Several cardiac genes contain glucocorticoid response elements in their promoter regions that confer transcriptional-level glucocorticoid responsiveness.22 Such genes include those that encode voltage-gated potassium channels, as well as protein kinases, which serve to phosphorylate and regulate the voltage-gated sodium channels. This expression may be chamber specific and might play a role in the developing fetal heart. In addition, there are more rapidly acting, nontranscriptional pathways by which cortisol can regulate the activity of voltage-gated potassium channels.
The cardiac effects of glucocorticoid excess in Cushing disease arise from the effects of glucocorticoids on the heart, liver, skeletal muscle, and fat tissue.22–24 Accelerated atherosclerosis can result from abnormal glucose metabolism with hyperglycemia, hyperinsulinemia, and hypertension in most patients, as well as from altered clotting and platelet function.22 The mechanism of cortisol-mediated hypertension is multifactorial. In contrast to aldosterone-induced hypertension, intracranial administration of glucocorticoids lowers blood pressure.25 Thus cortisol-mediated hypertension appears not to result from activation of the mineralocorticoid receptor. In addition, antagonism of the effects of glucocorticoid via its cytosolic receptor can block the cortisol-induced elevations in glucose and insulin levels, but not those related to blood pressure.26,27 Interestingly, one study suggested that inhibition of sodium retention is also insufficient to block the cortisol-mediated rise in blood pressure, thus pointing to changes in vascular reactivity, systemic vascular resistance, and nitric oxide–mediated vasodilation as candidates for the hypertensive effect.22
The rise in serum glucose levels and the development of insulin resistance may activate proinflammatory cytokines such as tumor necrosis factor-alpha and interleukin-6 (IL-6), which may underlie the accelerated atherosclerosis of insulin resistance found in other endocrine disease states.26 Although it typically acts as an anti-inflammatory hormone, cortisol excess can promote inflammation and accelerate atherosclerosis by producing insulin resistance, changes in corticosteroid binding protein, and regulation of proinflammatory cytokines.28 The centripetal obesity characteristic of glucocorticoid excess resembles that seen in insulin resistance (metabolic) syndromes. The excess androgen production resulting from increased stimulation of the adrenal cortex by ACTH may also accelerate atherosclerosis in both men and women.
The increased cardiovascular morbidity and mortality of Cushing syndrome can be explained largely by cerebrovascular disease, peripheral vascular disease, coronary artery disease with myocardial infarction, and chronic congestive heart failure.24,27 Studies of left ventricular structure and function have shown hypertrophy and impaired contractility in 40% of patients.29 Cushing syndrome can be manifested as dilated cardiomyopathy.30 A recent study of 15 patients with Cushing syndrome showed increased coronary artery calcification and plaque volume, as measured by Agatston scores, when compared with age-matched controls.31 In addition, the marked muscle weakness resulting from corticosteroid-induced skeletal myopathy contributes to impaired exercise tolerance.
Patients with Cushing disease can exhibit a variety of electrocardiographic changes. The duration of the PR interval appears to correlate inversely with adrenal cortisol production rates.22 The mechanism underlying this correlation may be related to expression or regulation of the voltage-gated sodium channel (SCN5A). Changes on the ECG, specifically in the PR and QT intervals, may also arise from the direct (nongenomic) effects of glucocorticoids on the voltage-gated potassium channel (Kv1.5) in excitable tissues.22,24
A particular complex of cardiac and adrenal lesions, referred to as the Carney complex, combines Cushing syndrome, cardiac myxoma, and a variety of pigmented dermal lesions (not café au lait spots). This monogenic autosomal dominant trait maps to the q2 region of chromosome 17.32 Myxomas most commonly occur in the left atrium but can arise throughout the heart, can develop at young ages, and can be multicentric.
Diagnosis
Diagnosis of Cushing disease and Cushing syndrome requires the demonstration of increased cortisol production as reflected by an elevated 24-hour urinary free cortisol or nocturnal salivary cortisol level.28 ACTH measurements to determine whether the disease is pituitary, adrenal, or ectopically based and anatomic localization of the suspected lesions with MRI confirm the laboratory findings. Except in the setting of childhood asthma or other diseases requiring high-dose corticosteroid therapy, Cushing syndrome is rare in the pediatric population.
Treatment
Treatment of excess cortisol production depends on the underlying mechanisms. In Cushing disease, transsphenoidal hypophysectomy with or without postoperative radiation therapy can partially or completely reverse the increased ACTH production by the anterior pituitary. Cushing syndrome requires surgical removal of one (adrenal adenoma, adrenal carcinoma) or both (multiple nodular) adrenal glands. In nonsurgical patients, the adrenal enzyme inhibitor ketoconazole can reverse the excessive cortisol production. Immediately after surgery, cortisol and mineralocorticoid (fludrocortisone) must be replaced to prevent adrenal insufficiency. Treatment of the ectopic ACTH syndrome requires identification and treatment of the neoplastic process. Clinical signs and symptoms of Cushing syndrome often develop in patients treated with exogenous steroids at doses equivalent to 20 mg of prednisone daily for more than 1 month. Even mild or subclinical degrees of Cushing syndrome (adrenal incidentaloma) appear to increase the risk for cardiovascular disease.30 In all cases, treatment leading to remission of the hypercortisolism led to amelioration of the cardiac changes.33
Hyperaldosteronism (See also Chapter 43)
Aldosterone production by the zona glomerulosa is under control of the renin-angiotensin system. Renin secretion responds primarily to changes in intravascular volume. Aldosterone synthesis and secretion are primarily regulated by angiotensin II, which binds to the angiotensin II type I receptor on cells of the zona glomerulosa.34
Aldosterone’s mechanism of action on target tissues resembles that reported for glucocorticoids (Fig. 81-1). Aldosterone enters cells and binds to the mineralocorticoid receptor, which then translocates to the nucleus and promotes the expression of aldosterone-responsive genes. In addition to kidney cells, in which mineralocorticoid receptors control sodium transport, in vitro studies have demonstrated these receptors in rat cardiac myocytes; they respond to mineralocorticoid stimulation with an increase in protein synthesis. Whether these changes correspond to any relevant in vivo cardiac effects is unclear, but aldosterone may augment the development of cardiac hypertrophy and diastolic dysfunction in patients with hypertension.35
The aldosterone antagonists spironolactone and eplerenone compete for receptor binding in the cytosol (Fig. 81-1). In addition to the treatment of primary hyperaldosteronism, recent studies have defined a role for these agents after acute myocardial infarction and for the treatment of left ventricular dysfunction, heart failure, and hypertension (see Chapters 25 and 44).36,37
Although the major cause of increased serum aldosterone levels is related to the physiologic response to activation of the renin-angiotensin system, well-recognized aldosterone-producing benign adrenal adenomas can occur (Conn syndrome). Primary hyperaldosteronism augments sodium retention, causes hypertension, increases renal loss of magnesium and potassium, decreases arterial compliance with a rise in systemic vascular resistance and subsequent vascular damage, and alters sympathetic and parasympathetic neural regulation.38 Many of the changes in the heart and cardiovascular system in hyperaldosteronism result from the associated hypertension.35 Primary aldosteronism may promote atrial fibrillation in patients with hypertension but no structural heart disease. A recent review discussed the approach to the detection, diagnosis, and treatment of patients with primary aldosteronism.39 The genetics of this clinical syndrome and the mechanisms underlying the cardiovascular sequelae, including modulation of the KCNJ5 gene, remain a focus of study.40 The hyperaldosterone-mediated hypokalemia and much of the associated hypertension respond to surgical removal of a unilateral (or occasionally bilateral) benign adrenal adenoma.38
Addison Disease
Long before recognition that the glands situated just above the upper pole of each kidney (suprarenal) synthesize and secrete glucocorticoids and mineralocorticoids, Thomas Addison described the association of atrophy and loss of function of these structures with marked changes in the cardiovascular system. Acute addisonian crisis, one of the most severe endocrine emergencies, is characterized by hypovolemia, hypotension, and acute cardiovascular collapse resulting from renal sodium wasting, hyperkalemia, and loss of vascular tone. Adrenal insufficiency arises most commonly from bilateral loss of adrenal function on an autoimmune basis; as a result of infection, hemorrhage, or metastatic malignancy; or in selected cases, from inborn errors of steroid hormone metabolism.41 In contrast, secondary adrenal insufficiency, which results from pituitary-dependent loss of ACTH secretion, leads to a fall in glucocorticoid production, whereas mineralocorticoid production, including aldosterone, remains at relatively normal levels. Studies have addressed the issue of relative hypothalamic-pituitary-adrenal insufficiency in acutely ill patients. Although the actual existence of such an entity and diagnostic criteria for establishing this condition remain to be validated, it has reopened the question of the need for stress-dose cortisol treatment in the management of patients with critical illness.42
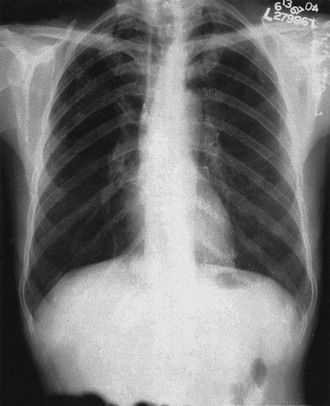
Addison disease can occur at any age. In children it may be associated with autoimmune (Hashimoto) thyroid disease. The noncardiac symptoms—including increased pigmentation, abdominal pain with nausea and vomiting, and weight loss—can be chronic, but tachycardia, hypotension, loss of autonomic tone, and electrolyte abnormalities herald impending cardiovascular collapse and crisis.41 Blood pressure measurements uniformly show low diastolic pressure (<60 mm Hg) along with orthostatic changes that reflect loss of volume and acquired autonomic dysfunction. Laboratory findings in patients with hyponatremia and hyperkalemia indicate loss of aldosterone production (renin levels are high). Hyperkalemia can alter findings on the ECG by producing low-amplitude P waves and peaked T waves. Patients with newly diagnosed, untreated Addison disease have reduced left ventricular, end-systolic, and end-diastolic dimensions in comparison to controls. Cardiac atrophy is an unusual condition; it is seen with malnutrition caused by anorexia, in astronauts after prolonged space flight, in populations with sodium-deficient diets, and characteristically with Addison disease (teardrop heart; Fig. 81-2). This atrophy reflects a response to decreases in cardiac workload because restoration of normal plasma volume with mineralocorticoid and glucocorticoid replacement increases ventricular mass.
Diagnosis
Acute adrenal insufficiency characteristically occurs in the setting of acute stress, infection, or trauma in a patient with chronic autoimmune adrenal insufficiency or in children with congenital abnormalities in cortisol metabolism. It can also develop as a result of bilateral adrenal hemorrhage in patients with severe systemic infection or diffuse intravascular coagulation.41 Secondary adrenal insufficiency can occur in the setting of hypopituitarism and is usually chronic, but acute changes caused by pituitary hemorrhage (apoplexy) or pituitary inflammation (lymphocytic hypophysitis) can also occur. Acute adrenal insufficiency can develop in patients treated long-term with suppressive doses of corticosteroids (>10 mg of prednisone for more than 1 month) if treatment is stopped precipitously or if an acute severe non–endocrine-related illness arises.
The diagnosis is established when cortisol levels are low (<8 mg/dL) in the morning or during severe stress and fail to rise higher than 20 mg/dL 30 minutes after an intravenous (IV) injection of 0.25 mg of cosyntropin. Diagnosis in the setting of acute illness may be more difficult, and a low (<10 mg/dL) morning serum level of cortisol may suffice to suggest impaired control of secretion.42
Treatment
Management of acute addisonian crisis needs to address three major issues. The first is adequate hydrocortisone replacement—100 mg given as an initial IV bolus, then 100 mg every 8 hours for the first 24 hours, and finally tapering of the dose over the next 72 to 96 hours. The second is restoration of the intravascular fluid deficit with large volumes of normal saline with 5% dextrose. The third is identifying and treating any underlying precipitating cause, including infection, acute cardiac or cerebral ischemia, or intra-abdominal emergency. Long-term treatment consists of oral corticosteroid and mineralocorticoid (fludrocortisone, 0.1 mg/day) replacement, but these patients are at increased risk for all-cause and cardiovascular mortality.43
Prolactin Disease
The most common disorder of the anterior pituitary gland is small (<1.0 cm), prolactin-producing pituitary adenomas causing amenorrhea and galactorrhea. Because prolactin secretion is normally inhibited by hypothalamic dopamine, dopamine agonists such as cabergoline and bromocriptine are first-line treatments. Unlike the experience with Parkinson disease, however, such treatment has not been linked with cardiac valvular disease.44
Parathyroid Disease
Diseases of the parathyroid glands can produce cardiovascular disease and alter cardiac function through two mechanisms. The first is changes in the secretion of parathyroid hormone (PTH), a protein hormone that affects the heart, vascular smooth muscle cells, and endothelial cells. The second mechanism is changes in serum calcium levels. Serum ionized calcium regulates the synthesis and secretion of PTH by an exquisitely sensitive negative feedback mechanism.45
PTH can bind to its receptor and alter the spontaneous beating rate of neonatal cardiac myocytes through an increase in intracellular cyclic adenosine monophosphate (cAMP). PTH can also alter calcium influx and cardiac contractility in adult cardiac myocytes and relaxation of vascular smooth muscle cells. Moreover, the structurally related PTH-related peptide (PTHrP) is synthesized and secreted in a variety of tissues, including cardiac myocytes. PTHrP can bind to the PTH receptor on cardiac cells and stimulate accumulation of cAMP and contractile activity, as well as regulate L-type calcium currents. Thus the direct effects of increased serum levels of PTHrP on the heart and systemic vasculature can accompany paraneoplastic syndromes characterized by hypercalcemia. Long-term treatment with the recombinant human PTH teriparatide (Fortéo) may require monitoring for adverse cardiac effects.
Hyperparathyroidism
The classic primary hyperparathyroidism producing hypercalcemia most often results from adenomatous enlargement of one of the four parathyroid glands. Cardiovascular actions of hypercalcemia include an increase in cardiac contractility; shortening of the ventricular action potential duration, primarily through changes in phase 2; and blunting of the T wave and changes in the ST segment, occasionally suggesting cardiac ischemia.45 The QT interval is shortened and occasionally accompanied by decreases in the PR interval. Treatment with digitalis glycosides appears to increase sensitivity of the heart to hypercalcemia.
Hypercalcemia may lead to pathologic changes in the heart, including the myocardial interstitium and conducting system, as well as calcific deposits in the valve cusps and annuli. Although initially observed in fairly longstanding and severe hypercalcemia, so-called metastatic calcifications can also occur in secondary parathyroid disease arising from chronic renal failure, in which the serum calcium-phosphorus product constant is exceeded. Left ventricular systolic function is generally maintained in patients with primary hyperparathyroidism, but severe or chronic disease may impair diastolic function. Changes in left ventricular structure and function do not appear to improve by 1 to 2 years after successful parathyroid surgery.45,46
A simultaneous increase in serum immunoreactive PTH (best represented by the intact PTH assay) with elevation of the serum calcium level establishes the diagnosis of primary hyperparathyroidism. Other causes include hypercalcemia of malignancy with an increased level of PTHrP or hypercalcemia arising directly from bony metastases or neoplastic (lymphoma) or non-neoplastic (sarcoidosis) disease leading to an increase in the synthesis and release of 1,25-dihydroxyvitamin D3. Treatment of hyperparathyroidism is surgical removal of the parathyroid adenoma.46
Hypocalcemia
Low serum levels of total and ionized calcium directly alter myocyte function. Hypocalcemia prolongs phase 2 of the action potential duration and the QT interval. Severe hypocalcemia can impair cardiac contractility and gives rise to a diffuse musculoskeletal syndrome consisting of tetany and rhabdomyolysis. Primary hypoparathyroidism is rare and can develop after surgical removal of the parathyroid glands, as may occur after treatment of thyroid cancer, in the setting of polyglandular dysfunction syndromes, as a result of glandular agenesis (DiGeorge) syndrome, and in the rare heritable disorder pseudohypoparathyroidism.
Chronic renal failure is the most common cause of low serum calcium and high PTH levels. In such patients the effects of chronically high levels of PTH (secondary hyperparathyroidism) on the heart and cardiovascular system may both be causative and serve as a biomarker in assessing heart failure treatment strategies.47,48 In elderly patients with progression of aortic stenosis, a rise in serum PTH and bone remodeling occurs.49 The ability of PTH to stimulate G protein–coupled receptors may impair myocyte contractility and contribute to LVH. Cinacalcet, a recently approved calcimimetic agent, can be used to treat the secondary hyperparathyroidism associated with chronic renal failure. A trial to assess its effectiveness on cardiovascular events, however, showed no significant benefit.50
Vitamin D
Observational evidence suggests that lower levels of vitamin D (<30 ng/mL of 25-hydroxyvitamin D) are associated with increased all-cause and cardiovascular morbidity.51 In postmenopausal women, increased vitamin D intake reduces their relative risk for the development of cancer. Although low levels of vitamin D occur in patients with chronic renal disease and heart failure, we await the results of ongoing trials to evaluate whether vitamin D supplementation might prevent cardiac disease.52
Thyroid Gland
The thyroid gland and the heart share a close relationship that arises in embryology. In ontogeny, the thyroid and heart anlage migrate together. The close physiologic relationship is affirmed by predictable changes in cardiovascular function across the entire range of thyroid disease states; cardiovascular manifestations are some of the most common and characteristic findings of hyperthyroidism. Diagnosis and management of thyroid hormone–mediated cardiac disease states require understanding of the cellular mechanisms of thyroid hormone on the heart and vascular smooth muscle cells.53
Cellular Mechanisms of Thyroid Hormone Action on the Heart
Under the regulation of thyroid-stimulating hormone (thyrotropin, TSH), the thyroid gland concentrates iodide and, through a series of enzymatic steps, synthesizes predominantly tetraiodothyronine (T4, 85%) and a smaller percentage of triiodothyronine (T3, 15%; Fig. 81-3). The major source of T3 synthesis is conversion by 5′-monodeiodination, primarily in the liver, skeletal muscle, and kidneys. Studies have confirmed T3 as the active form of thyroid hormone that accounts for the vast majority of biologic effects, including stimulation of tissue thermogenesis, alterations in the expression of various cellular proteins, and actions on the heart and vascular smooth muscle cells.53,54 Free T3 enters cells via transport proteins (Fig. 81-4) of the MCT and OAT family of cell surface transporters.55 Most data indicate that cardiac myocytes cannot metabolize T4 to T3. Therefore despite the presence of the relevant enzymes, all the observed nuclear actions and changes in gene expression result from changes in blood levels of T3
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


